
Abstract
Bovine leukemia virus (BLV) causes a persistent infection with provirus formation in B-lymphocytes. A real-time polymerase chain reaction (PCR) based on the conserved BLV polymerase (BLV pol) gene sequences was developed. Dually labeled probes were used to permit detection by the 5′ exonuclease assay. The assay was validated with 350 samples of bovine peripheral blood mononuclear cells including 144 samples from BLV-seropositive animals worldwide (South America, Europe, Middle East, Australia) representing 5 of the recently described 7 BLV envelope–based genotypes. The BLV pol real-time PCR proved to be highly specific and sensitive with the detection of up to 1 copy of an internal control plasmid. The 95% confidence intervals for assay sensitivity and specificity were ≥98.27% and ≥98.33%, respectively. Restriction fragment length polymorphism and phylogenetic BLV pol–based sequence analysis of the investigated samples were performed and compared with the previous described BLV env–based genotypes. Grouping of the sequences based on the pol gene yielded similar results as the env gene–based assay.
Introduction
Bovine leukemia virus (BLV; family Retroviridae, subfamily Orthoretrovirinae, genus Deltaretrovirus) is an exogenous retrovirus that is structurally and functionally related to the Primate T-lymphotropic virus 1, 2, and 3 (PTLV-1, -2, -3) and Human T-lymphotropic virus 1 and 2 (HTLV-1 and -2). Bovine leukemia virus in cattle causes a persistent infection with virus replication in B lymphocytes. 3 The infection is transmitted horizontally through the transfer of infected cells via direct contact, through milk, and possibly by blood-sucking insects.16,17,34 The majority of the infected animals remain asymptomatic. In these asymptomatic infected animals, fewer than 1% of peripheral blood mononuclear cells (PBMCs) and up to 10% of the circulating B lymphocytes are found to be infected.17,23 In 20–30% of the animals, however, a permanent increase in the number of B lymphocytes in the peripheral blood can be observed. Persistent lymphocytosis (PL) results from the polyclonal expansion of untransformed lymphocytes, especially immunoglobulin (Ig)M+ and cluster of differentiation (CD)5+ B cells.7,22 In animals with PL, the majority (>60%) of the CD5+ B cells in the peripheral blood contain provirus. 23 Clinical signs of malignant lymphoma become evident as tumors invade different lymphoid tissues and can include enlarged lymph nodes, weight loss, decreased milk production, fever, and loss of appetite.
Bovine leukemia virus is highly prevalent in many countries worldwide11,17 and has a significant economic impact on the dairy cattle industry mainly due to reduced milk production, increased replacement costs, reduced conception rates, and a greater susceptibility to other infectious diseases such as mastitis, diarrhea, and pneumonia.6,9,12,28,29,37,38 Over the last 3 decades, many European countries have emphasized the eradication of enzootic bovine leukosis, resulting in eradication (≥99.8% farm seronegative rate) in countries such as Austria, Belgium, Czech Republic, Denmark, Finland, France, Germany, Ireland, Luxembourg, The Netherlands, Norway, Slovakia, Slovenia, Spain, Sweden, United Kingdom, and certain administrative regions in Italy, Poland, and Portugal. In spite of these achievements, some ambiguous serology results are still encountered, with occasional occurrence of seronegative but polymerase chain reaction (PCR)-positive samples.1-3,13,20 Analysis of the BLV envelope (env) gene collected in multiple geographical locations demonstrated significant sequence conservation. 5 Nevertheless, up to 7 genotypes were identified by BLV env–based restriction fragment length polymorphism (RFLP) analysis.5,13 Whereas genotypes A and C–G can be reliably detected with serological methods such as agar gel immunodiffusion test and enzyme-linked immunosorbent assays (ELISAs), genotype B is sometimes undetectable or only transiently detectable by these methods.13,18 Classical PCR and loop-mediated isothermal amplification (LAMP) methods based on conserved group-specific antigen (gag) and env gene sequences have been described for the detection of provirus in PBMC, milk, and tissue samples from infected animals.4,13-15,20,21,27,36 The necessity to perform most of the PCR tests in a nested format to achieve adequate sensitivity, however, also increases the likelihood for laboratory cross-contaminations. The LAMP method proved to be robust but did not detect all BLV-infected samples. 20 To further improve the detection of BLV-infected cattle, a TaqMan real-time PCR assay was developed based on the conserved BLV polymerase (pol) gene and validated using 350 archived DNA samples collected from South America, Europe, Middle East, and Australia.
Materials and methods
Identification of target sequences and generation of a control plasmid
Sequence analysis was performed from all BLV full-length pol genes available through the National Center for Biotechnology Information (NCBI; AF033818, M10987, EF600696, K02120, D00647, AF257515, NC_001414, FJ914764). a Alignment-based primer and probe selection (Table 1) was supported by a software package. b All oligonucleotides and the control plasmid were synthesized by a commercial company c and stored at −20°C until use. The probes were 5′-end labeled with 6-carboxyfluorescein (FAM) and 3′-end labeled with black hole quencher 1 to permit real-time PCR detection by the 5′ exonuclease assay.
Primers and probes used in the current study.

Nucleotide positions according to GenBank accession no. AF033818. 31
5′-end of the probes was labeled with 6-carboxyfluorescein (FAM), and the 3′-end was labeled with black hole quencher 1 (BHQ1).
5′-end of the probe was labeled with hexachloro-6-carboxyfluorescein (HEX), and the 3′-end was labeled with BHQ1.
A control plasmid was constructed for the BLV pol real-time PCR to verify the assay sensitivity and to have a positive control that can easily be distinguished from the wild-type DNA. For this purpose, a 112-bp fragment corresponding to the BLV pol real-time target sequence was cloned into plasmid pCR2.1. c The sequence is identical with nucleotides (nt) 3088–3205 (AF033818) except for the BLV pol control probe binding region between nt 3153 and 3176. The control probe binding region consists of the nucleotides for the BLV pol probe binding region (nt 3116–3139) in a randomized fashion (Table 1) in order to enable the design of a hexachloro-6-carboxyfluorescein (HEX)-labeled BLV pol control probe that binds the corresponding target sequence of the plasmid with similar reaction kinetics such as the FAM-labeled BLV pol probe for the wild-type sequence. Therefore, FAM- and HEX-labeled probes bind to the control plasmid (pCR2.1-BLV-pol), whereas the FAM-labeled probe binds only to BLV-positive wild-type sequences.
The concentration of the plasmid was determined spectrophotometrically, and the exact number of plasmid molecules was calculated using the formula [X (g/µl DNA)/(length in nucleotides × 660)] × 6.022 × 1023 = number of molecules/µl. The lower detection limit of the real-time PCR was determined by amplifying serial 10-fold dilutions of the plasmid pCR2.1-BLV-pol in TE (10 mM Tris-HCl [pH 8.0], 1 mM ethylenediamine tetra-acetic acid [EDTA]) buffer containing 100 ng/µl of BLV-negative genomic DNA. The stocks of the plasmid were stored at −70°C.
In addition to the control plasmid, DNA from persistently BLV-infected PO714 cells was used to investigate assay sensitivity by serial dilution of DNA equivalent to 1,000, 100, 10, 1, 0.75, 0.5, 0.25, and 0.1 cells in 100 ng/µl of DNA derived from the BLV-negative cell line PO. The dilutions performed in triplicate were subsequently investigated by real-time PCR.
Samples and DNA isolation
A total of 350 bovine PBMC samples were analyzed. Ethylenediamine tetra-acetic acid–blood samples were collected from 144 serologically positive animals (Table 2), which included 133 samples from naturally infected animals from Europe (Croatia, Germany, Poland, Russia, Slovenia, Sweden, Ukraine), Argentina, New Zealand, and Saudi Arabia and 11 samples from experimentally BLV-infected cattle in Germany. A second set of EDTA-blood samples were derived from 168 animals in Europe (Croatia, Germany, Poland, Slovenia, Sweden) and Argentina. No serological test could be performed from 38 samples from Brazil and Egypt as only PBMC-derived DNA had been obtained (Table 2).
Samples analyzed with the Bovine leukemia virus (BLV) env gene nested polymerase chain reaction (PCR) and the real-time BLV pol PCR.*
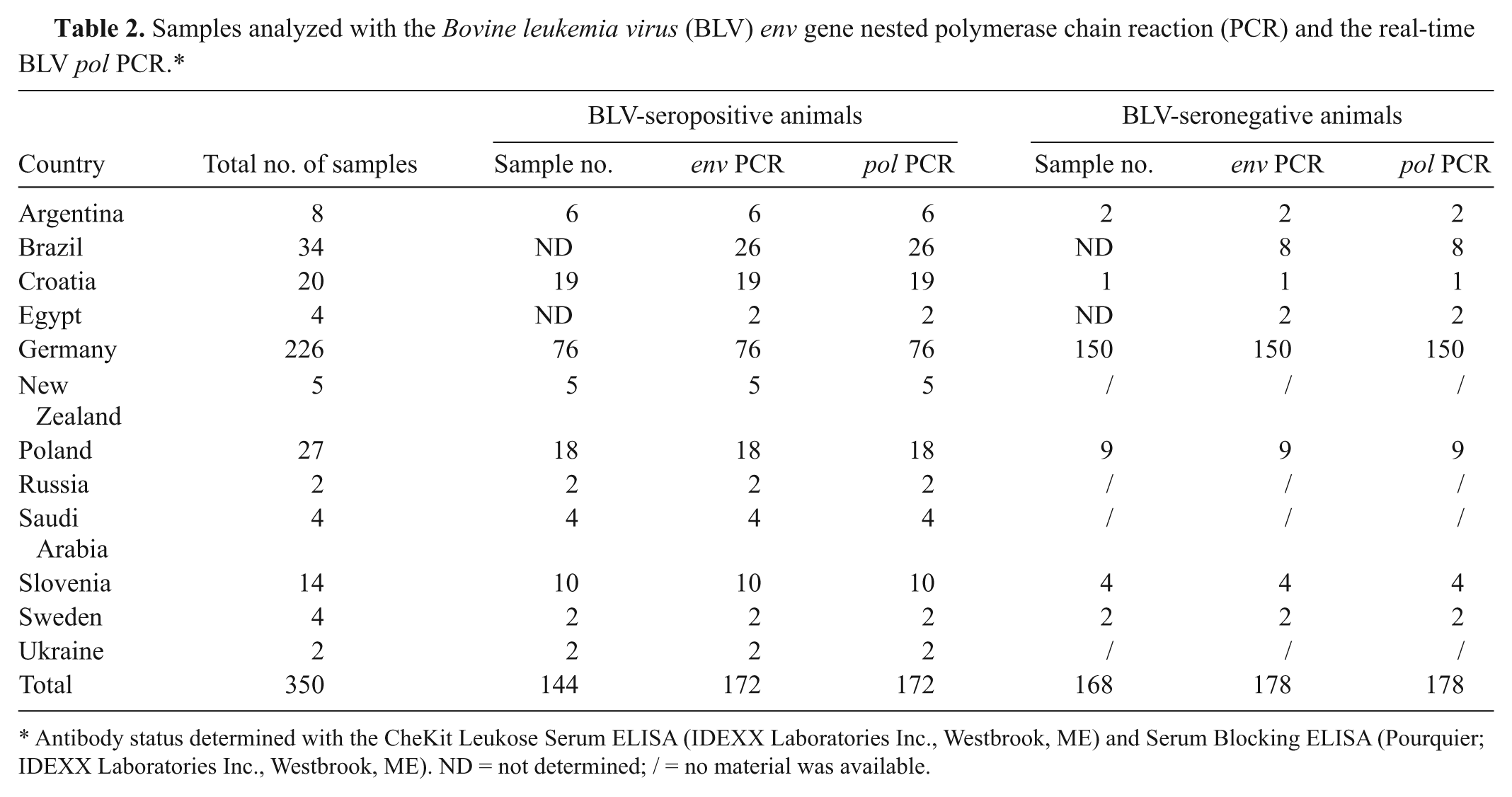
Antibody status determined with the CheKit Leukose Serum ELISA (IDEXX Laboratories Inc., Westbrook, ME) and Serum Blocking ELISA (Pourquier; IDEXX Laboratories Inc., Westbrook, ME). ND = not determined; / = no material was available.
Cell lines persistently infected with BLV (PO714), Bovine immunodeficiency virus (BIV; FBS-BIV 198), and Bovine foamy virus (BFV; BHK21-C13) were used to exclude cross-reactivity with other bovine retroviruses. For Jembrana disease virus (JDV), a sequence comparison with the BLV-specific primers and probes was performed.
Peripheral blood mononuclear cells were isolated from EDTA-blood samples using density gradient d centrifugation according to the manufacturer’s instruction. DNA isolation from the PBMC and cell lines was performed using a commercial kit e following the manufacturer’s instructions. The amount of DNA was measured spectrophotometrically f and by gel electrophoresis; 0.5 µg was used in the BLV env and BLV pol PCR. The DNA samples were stored at −70°C.
Real-time PCR based on the conserved BLV pol gene
The PCR was performed using a 20-µl reaction volume and the TaqMan universal PCR master mix g including AmpliTaq Gold DNA Polymerase, AMPErase UNG, deoxyribonucleotide triphosphate with 2′-deoxyuridine 5′-triphosphate, and passive reference dye (5-carboxy-X-rhodamine [ROX]). For one reaction, the assay was optimized to 10 µl of universal PCR master mix, 2 µl of DNA, 0.6 µl each of the pol-specific primers (10 pmol/µl BLV-pol-s, 10 pmol/µl BLV-pol-as), 0.3 µl of BLV pol probe (10 pmol/µl), 0.3 µl of BLV pol internal control probe (10 pmol/µl), and 6.2 µl of distilled H2O. The positive control contained 104 molecules of the control plasmid. The reaction was carried out using a real-time PCR system h with the following temperature profile: 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 58°C. Fluorescence data were collected during the annealing step. Data were analyzed with MXPro4 software i using the dR(n)-mode, blocking background fluorescence and neutralizing data to the reference dye (ROX) standard.
BLV env nested PCR, RFLP analysis, and ELISA
Using the primers BLV-env-s (5′-TCTGTGCCAAG TCTCCCAGATA-3′; nt 5032–5053) and BLV-env-as (5′-AACAACAACCTCTGGGAAGGGT-3′; nt 5608–5629), as well as BLV-env-nested-s (5′-CCCACAAGGGCGG CGCCGGTTT-3′; nt 5099–5021) and BLV-env-nested-as (5′-GCGAGGCCGGGTCCAGAGCTGG-3′; nt 5521–5542), a 444-bp fragment of the env gene was amplified (using the conditions described previously13,14). j The obtained 444-bp PCR products were digested with BamHI, PvuII, and BclI k and analyzed by gel electrophoresis in order to determine the BLV subtypes according to RFLP analysis.13,14 The antibody status of the sera was determined with a commercial ELISA l following the manufacturer’s instructions.
Results
Identification of target sequences
The comparative sequence analysis performed with the available full-length BLV pol sequences in parallel with the search for appropriate primer/probe combinations revealed that within the pol gene, a 113-bp region spanning nt 3093–3205 (nt according to AF033818) could be suited for a real-time PCR due to a high homology with the previously published BLV pol sequences.5,10,30,35 Single nucleotide changes were noticed only in individual sequences (Table 3).
Sequence alignment of the Bovine leukemia virus (BLV) pol gene real-time polymerase chain reaction primer/probe combinations from peripheral blood mononuclear cell samples derived from all 5 BLV env gene clusters.*
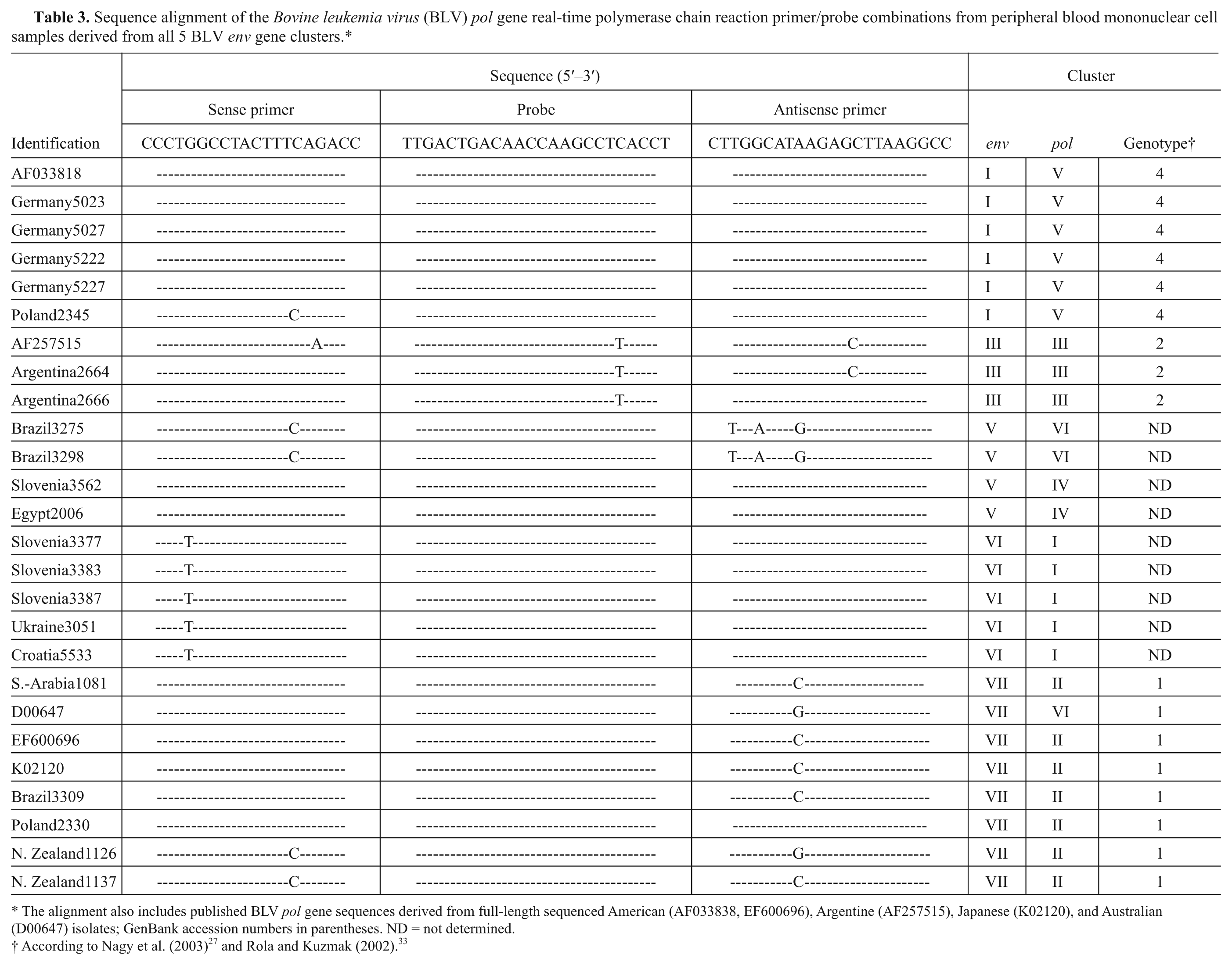
The alignment also includes published BLV pol gene sequences derived from full-length sequenced American (AF033838, EF600696), Argentine (AF257515), Japanese (K02120), and Australian (D00647) isolates; GenBank accession numbers in parentheses. ND = not determined.
As control plasmid, a 112-bp fragment (nt 3088–3205, AF033818) corresponding to the BLV pol real-time target sequence was cloned into plasmid pCR2.1. The plasmid sequence between nt 3153 and 3176 was randomized with regard to the BLV pol probe binding region (nt 3116–3139; Table 1) in order to enable the design of a BLV pol control probe, which binds the corresponding target sequence of the plasmid with similar reaction kinetics as the BLV pol probe for the wild-type sequence. The sequence of the constructed plasmid was verified by sequence analysis.
Sensitivity and specificity of the real-time BLV pol PCR
The annealing temperature of the real-time BLV pol PCR was verified to 58°C. Serial 10-fold dilution of the plasmid pCR2.1-BLV-pol revealed that the primers and the FAM- (BLV pol) and HEX- (BLV pol control) labeled probes bind with similar kinetics to the target sequence. Experiments using DNA from BLV-infected animals revealed that the wild-type sequence can only be detected with the FAM-labeled probe. Assay sensitivity using plasmid pCR2.1-BLV-pol revealed that the real-time PCR is able to detect 1–5 molecules of the target sequence with the FAM- as well as the HEX-labeled probe. The point estimate for sensitivity (Se) and specificity (Sp) was 100%. The one-sided confidence interval was used for calculating true assay sensitivity and specificity. With regard to the seropositive animals, the 95% confidence interval for the ELISA was 97.94% (Se ≥ 97.94%) and for the env and pol PCR 98.27% (Se ≥ 98.27%). For the seronegative animals, the 95% confidence interval for the ELISA was 98.23% (Sp ≥ 98.23) and for the env and pol PCR 98.33 (Sp ≥ 98.33).
For further assay analysis, serial dilutions of DNA derived from persistently infected PO714 cells in 100 ng/µl of BLV-negative DNA revealed that a DNA equivalent of one PO714 cell could still be detected. Fewer amounts of PO714 cell–derived DNA failed to reproducibly generate a positive reaction. Additional serial dilution experiments using DNA derived from PBMCs of a BLV-infected animal with PL showed that dilutions (in 100 ng/µl of BLV-negative DNA) containing 1 × 10−6 to 1 × 10−8 µg DNA were still positive.
To exclude the amplification of other bovine retroviruses, BIV- (FBS-BIV 198) and BFV- (BHK21-C13) infected cell cultures were investigated. The results proved that BIV and BFV proviral DNAs are not detected by the assay. Comparison of the primers and probes to the 2 published JDV sequences Pul01 and Kal07 revealed that no more than 5 bases are complementary, which makes it highly unlikely that the real-time PCR will amplify JDV proviral DNA.
Real-time BLV pol PCR
From 312 samples with known serological BLV status as determined by commercial ELISA, l all 144 samples derived from BLV-seropositive animals were detected by the real-time BLV pol PCR with threshold cycle (Ct) values ranging between 22 and 39. Twenty-six of the samples from Brazil and 2 of the samples from Egypt were also positive in the PCR (Table 2). All 168 BLV-seronegative animals were negative by the real-time BLV pol PCR with Ct values above 40. Eight of the samples from Brazil and 2 of the samples from Egypt were negative in the PCR (Table 2).
BLV env nested PCR and RFLP analysis
All DNA samples were also investigated with the BLV env nested PCR, which has been used previously as a reliable PCR method for BLV detection.13-15,36 Similar to the detection with the real-time PCR, the investigation using the BLV env nested PCR confirmed the presence and absence of proviral DNA in the same 172 and 178 samples, respectively, included in the study. The samples from Brazil and Egypt reacted similarly to the real-time PCR (Table 2).
From the 144 BLV-seropositive samples, 29 were selected for RFLP analysis. The results revealed that 5 of the 7 described RFLP-based BLV genotypes 13 were present in the sample collection. Ten samples belonged to variant A (Belgian subtype) as the 444-bp BLV env nested PCR products were cleaved with PvuII and BclII resulting in fragments of 280 bp/165 bp and 225 bp/220 bp length, respectively. Three samples were identified as variant B (Japanese subtype) with BamHI and BclI fragments of 315 bp/130 bp and 220 bp/120 bp/105 bp, respectively. Four samples belonged to variant C (Australian subtype) with BamHI and BclI fragments of 315 bp/130 bp and 225 bp/220 bp, respectively. One sample belonged to variant D with BclI fragments of 225 bp and 220 bp. Three samples belonged to variant F (Argentine subtype) with BamHI, PvuII, and BclI fragments of 315 bp/130 bp, 280 bp/165 bp, and 219 bp/225 bp, respectively. The RFLP variants E and G were not detected in the selected DNA samples.25,26,32
Discussion
The current study introduces a fast, sensitive, and specific method for the detection of BLV provirus sequences in PBMC of BLV-infected cattle. In contrast to most of the previous PCR assays that targeted the BLV gag or BLV env genes, the present study targeted conserved pol gene sequences suitable for a 5′ exonuclease-based real-time assay. The fact that the new real-time assay had an analytical sensitivity between 1 and 5 target molecules indicates that the assay is well suited for the current European BLV surveillance circumstance in which fewer than 1% of the PBMCs are usually found to be infected during the asymptomatic phase of infection. 17 In genetically major histocompatibility complex (MHC) class II BoLA-DRB3.2 allele–associated resistant cattle and BLV-infected animals with low or indeterminate humoral immune responses, proviral loads are usually even lower.8,19,24 This also explains why classic PCR assays frequently had to be performed in a nested format in order to obtain accurate results. In 2009, 20 a method based on the BLV-LTR region was described. Despite agreement between different serological and PCR-based detection systems in the majority of the 114 blood samples from BLV-infected farms, the LAMP results differed in 11 (approximately 10%) samples from serological test results and in 8 (approximately 7%) samples from a classical PCR method used. 20 The use of the control plasmid pCR2.1-BLV-pol with a unique “control” sequence in the present real-time PCR assay avoids problems with possible cross-contamination in routine use of the assay.
The lack of amplification using DNA from persistently BIV- and BFV-infected cells verified the specificity of the assay with regard to other bovine retroviruses. Low sequence homologies between the primer and probe sequences to JDV also seem to exclude the artificial detection of JDV with the BLV pol PCR. The concordant results between the real-time BLV pol PCR and the previously established BLV env nested PCR on the 350 archived DNA samples further validated the new assay.
Real-time PCR has many advantages over nested PCR. Real-time PCR is a shorter and less cumbersome procedure and is more amenable to high throughput testing. Furthermore, real-time PCR is less prone to cross-contamination. In summary, the described real-time method is suitable for detecting BLV provirus in infected animals.
Footnotes
Acknowledgements
The authors thank Kerstin Wink-Kruschke and Gabriele Czerwinski for technical support; Asia Gorska for the primer design; Martin Beer, Martin Groschup, and Anne Balkema-Buschmann for providing PBMC samples from BLV-negative animals; Roland Riebe for providing the cell lines PO714, FBS-BIV 198, and BHK21-C13; Mario Ziller for statistical analysis; and Shane Peterson, Murdoch University in Washington, for performing the sequence comparison analysis with regard to JDV.
a.
GCG Wisconsin software package, Accelrys Inc., San Diego, CA.
b.
Beacon Designer 2.06, PRIEMER Biosoft International, Palo Alto, CA.
c.
Eurofins MWG Operon Ebersberg, Bavaria, Germany.
d.
Histopaque-1077, Sigma-Aldrich, St. Louis, MO.
e.
QIAamp® DNA Blood Mini Kit, Qiagen, Hilden, North Rhine-Westphalia, Germany.
f.
Spectrophotometer DU 640 Beckman Coulter Inc., Fullerton, CA.
g.
TaqMan® universal PCR master mix, Applied Biosystems, Foster City, CA.
h.
Mx3000P real-time PCR system, Agilent Technologies Inc., Santa Clara, CA.
i.
MXPro4 software, Agilent Technologies Inc., Santa Clara, CA.
j.
Thermocycler Primus 25/96, MWG Biotech AG, Ebersberg, Bavaria, Germany.
k.
Restriction endonucleases, New England Biolabs, Ipswich, Suffolk, UK.
l.
CheKit Leukose Serum ELISA and Serum Blocking ELISA [Pourquier] by IDEXX Laboratories Inc., Westbrook, ME.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by the Federal Government of Germany and by the EU Network of Excellence, EPIZONE (contract no. FOOD-CT-2006-016236).