
Abstract
The current report discusses the process in which a methods comparison study in the National Animal Health Laboratory Network is performed. Specific details are provided for designing and analyzing studies intended to evaluate analytical sensitivity, efficiency, analytical specificity, cross-contamination, repeatability, operator variability, and to compare the performance of methods using diagnostic samples. As an example, a case study is presented comparing the performance of a candidate reverse transcription polymerase chain reaction (RT-PCR) chemistry to the current RT-PCR chemistry in use when the assay was originally validated. The present study revealed that, for all of the validation factors evaluated, the candidate method performed at least as well and generally better than the current method. The candidate method was, therefore, deemed fit for the original intended purpose of the current method and rendered acceptable for use. A discussion of the case study is intended to further motivate consideration of the study designs chosen.
Introduction
The National Animal Health Laboratory Network (NAHLN) is a laboratory network in the United States that employs standardized diagnostic protocols as part of its tenants. Prior to accepting an assay within the NAHLN, it must be rigorously validated in conjunction with World Organization for Animal Health (OIE) guidelines. 9 After validation, the accepted assay is deployed from the reference laboratory to the qualified NAHLN laboratories. Validated assays may require minor changes including, but not limited to, changes in PCR platform, reagents, or extraction methods. Minor changes are often dictated by the need for increased capacity in the event of an outbreak or due to discontinued or limited availability of reagents. The current report presents an approach in use by the NAHLN Methods Technical Working Group (MTWG) to assess the impact of minor modifications on fitness for intended purpose of the original validated assay. This approach provides a cost-efficient manner to evaluate the impact of a minor change of a validated assay while maintaining the NAHLN’s dedication to high quality standards. A de novo assay or an assay undergoing major changes would be subject to validation according to OIE guidelines prior to use within the NAHLN.
An example of a methods comparison study used by the NAHLN is presented. This example compares the candidate reverse transcription polymerase chain reaction (RT-PCR) chemistry to the current RT-PCR chemistry. This particular study was necessitated by the manufacturer’s discontinuation of the validated chemistry.
Stage I testing
Stage I testing is conducted to assess the analytical performance characteristics of an assay. 9 Generally, assay modification evaluations require a direct comparison with the current method while evaluations that apply only to the modification, such as assessing potential cross-contamination of newly introduced robotic equipment, does not require a direct comparison with the validated method.
Analytical sensitivity
The NAHLN MTWG follows the convention that defines the limit of detection (LOD) as the lowest concentration of analyte at which 95% of samples for that concentration are classified as positive.2,4 The NAHLN MTWG uses the lowest concentration in the dilution series where all replicates test positive as the estimate of the LOD rather than determining the 95% endpoint from a model fit.
To prepare for the LOD study, the analyte is dispensed into single-use aliquots. If the minor change relates to amplification, a pool of multiple extractions is required to achieve sufficient volume of extracted nucleic acid for the study. If the study is intended for evaluation of the extraction process, extraction is performed individually on each aliquot.
The concentration of the undiluted preparation is determined and expressed by any of several methods. Because the candidate and current methods are directly compared, the concentration may be expressed in terms of median tissue culture infectious dose (TCID50), median egg infectious dose (EID50), number of plaque-forming units, or genomic copy number. With the remaining bulk material, a dilution sequence is created using a 10-fold dilution factor. The volume at each dilution must be sufficient to create 6 aliquots, each with sufficient volume to be tested using either the candidate or current method. Testing occurs on 3 separate, independent occasions by a single operator. On each occasion, 2 of the aliquoted dilution series are tested using the candidate and current method, respectively. Independence is achieved by testing at different times and preparing fresh master mix for each testing occasion.
Generally, the NAHLN MTWG tests a minimum of 3 strains of an agent representing different serotypes, clades, or antigenic groups. Strain selection may be limited to 1 or 2 for agents with minimal variability or increased for agents with more diversity.
Case study—analytical sensitivity
Three isolates (A22 Iraq, O Rey, and SAT 3/2 South Africa 59) representing different serotypes of Foot-and-mouth disease virus (FMDV) were tested in the LOD experiment. For each isolate, 14 RNA extractions, using a commercially available kit, were performed with extracted RNA pooled prior to testing. Multiple extractions were necessary to obtain a sufficient volume of material to perform the experiment. A ten-fold dilution series (undiluted to10−9) of the RNA was made in 1× Tris–ethylenediamine tetra-acetic acid buffer (pH 8.0), with 1% carrier RNA. Individual, 4-μl aliquots were made to ensure RNA was thawed only once, used in the assay, and then discarded. One aliquot of each dilution for each isolate was tested on each of 3 separate real-time RT-PCR runs.
Figure 1 provides plots of the threshold cycle (Ct) values against log10 concentration for each isolate tested. The top row of plots depicts results obtained using the candidate chemistry and the bottom row using the current chemistry. Table 1 presents the LODs (expressed as log10 TCID50/ml). The LOD data for the candidate chemistry indicate at least as good or better analytical sensitivity among the 3 isolates tested than those for the current chemistry.
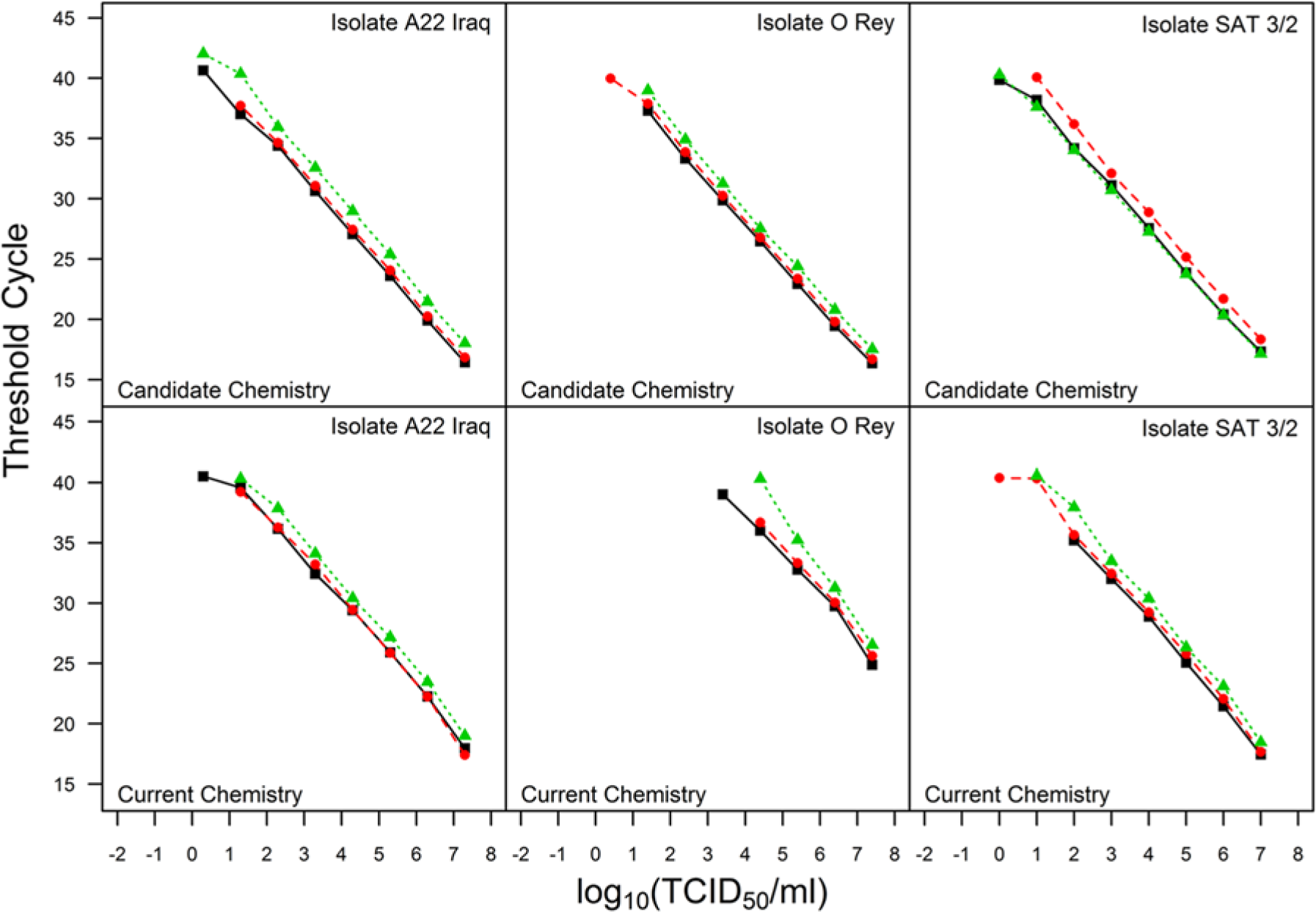
Limit of detection data where each plot displays threshold cycle versus log concentration for a unique Foot-and-mouth disease virus isolate and chemistry combination. The different colors and plotting characters represent testing that was conducted on different days.
Limit of detection (LOD; expressed as log10 TCID50/ml) and amplification efficiency (AE) estimates.*
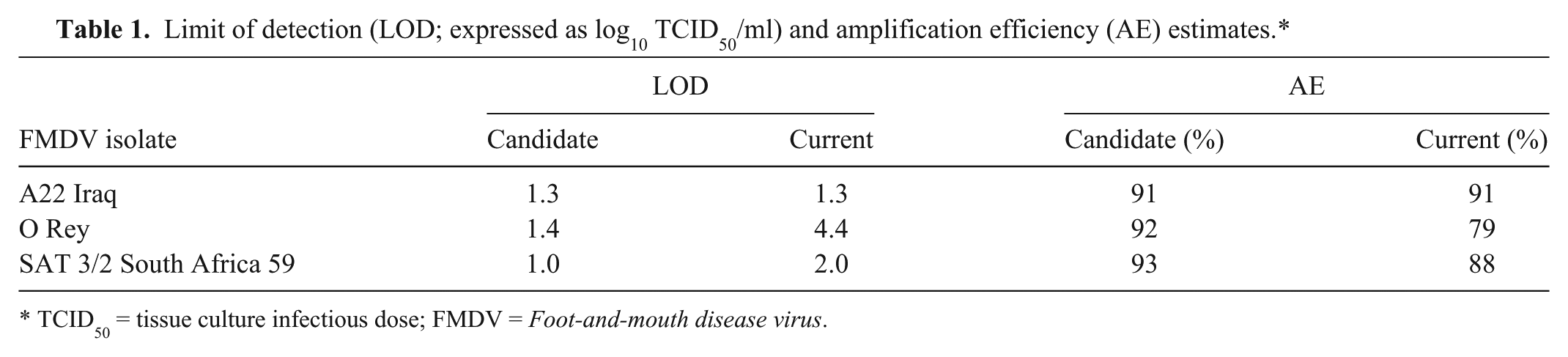
TCID50 = tissue culture infectious dose; FMDV = Foot-and-mouth disease virus.
Analytical specificity
Analytical specificity is generally defined as the ability of an assay to detect only the intended target and not related or potentially interfering nucleic acids or specimen-related artifacts. 2 At the time of validation, genetically similar organisms as well as other agents potentially found in a given matrix are tested for cross reactivity. Because a methods comparison study is recommended only for minor changes to the assay, retesting for analytical specificity may not be needed and was not performed in the case study. However, analytical specificity can be monitored by judiciously choosing diagnostic samples infected with potentially interfering nucleic acids during Stage II testing. Results of such an experiment should be presented in a table.
Efficiency
Data obtained from the LOD experiment are used to estimate the efficiency of the candidate and current method for each strain or isolate. The slope estimate from a linear regression of Ct on log10 concentration is used to estimate efficiency 4 as 100 × (10−1/slope – 1) using only those dilutions within the LOD.
Case study—efficiency
For the experiment conducted comparing the chemistries, the efficiency estimates were obtained using the mean slope from a random coefficients model fit to the data within the LOD from each chemistry and isolate combination separately. Table 1 shows that the efficiencies for the candidate assay were equal to, or better than, those of the current assay.
Repeatability
There are many sources of variability that contribute to the final test result. The current experiment was designed to estimate the sources of variability that are due to random differences between the same material within a run (intra-assay variability) and between runs (interassay variability) of the assay procedure. Others recommend expressing intra-assay variation by standard error bars or as confidence intervals on calibration curves. 4 Both measures express uncertainty in the mean rather than variances due to specific sources. The proposed alternative explicitly quantifies uncertainty within and between runs of the assay. A gauge repeatability and reproducibility (gauge R&R)3,8 study is proposed to estimate within-run (intra-assay variability) and between-run (interassay variability) variance.
The preparation of test material is dictated by the type of minor change being evaluated, and a study design is essential to obtain data suitable for estimating the quantities of interest. A single operator performs all testing using both the current and candidate method to assure the estimates only reflect the within- and between-run variance.
Two different options for preparing the material are presented. The first option is useful when the modification is related to the extraction of nucleic acid and the second option applies when the modification affects only its amplification (Fig. 2).
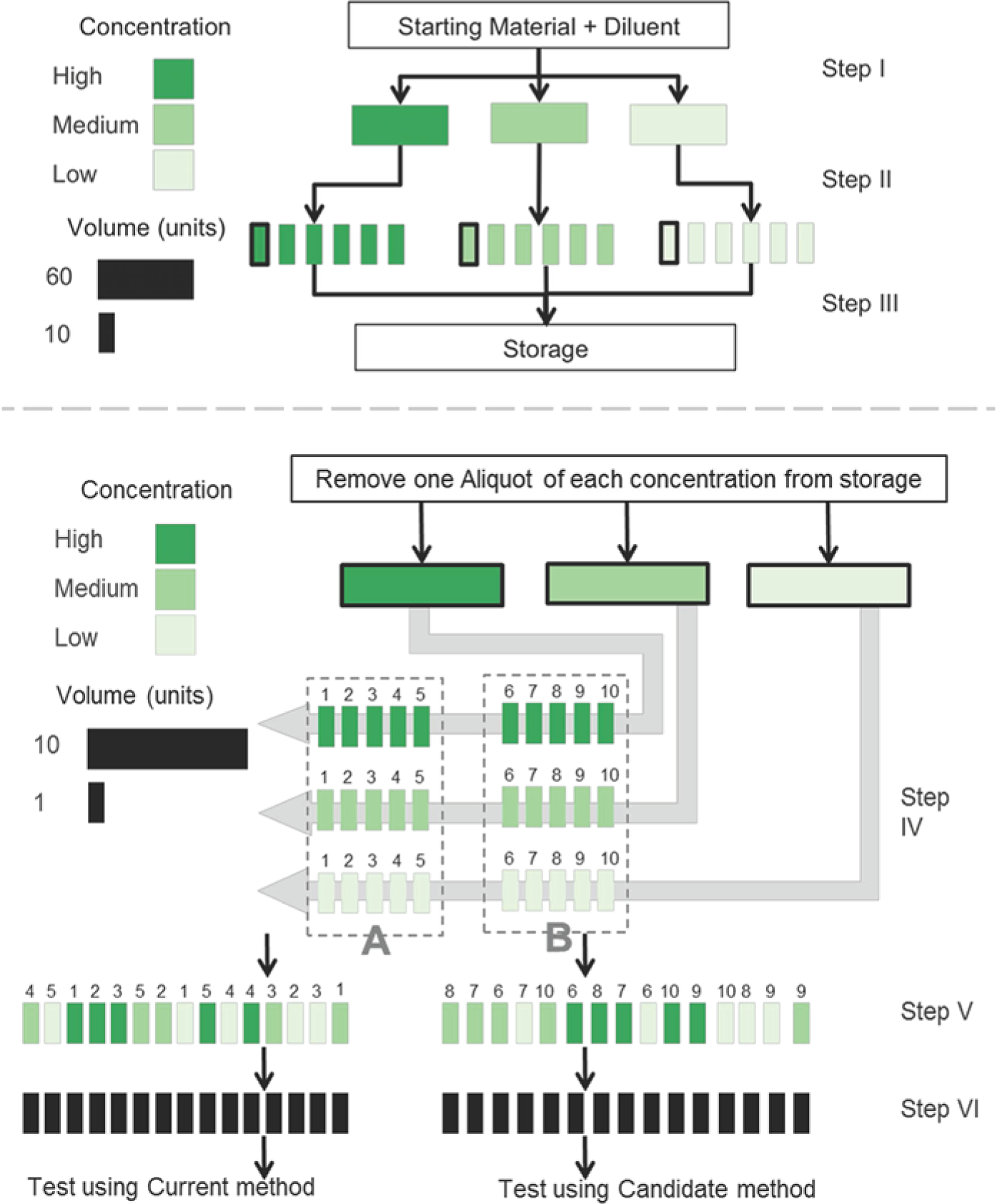
A visual depiction of preparing samples for the repeatability study when the modification under evaluation affects the extraction method. Dilutions are created (step I) and divided into aliquots (step II) and stored (step III) until use. At the time of testing, 1 aliquot of each dilution is removed from storage and further divided (step IV) for individual extractions and reactions for both the current method (A) and the proposed method (B). Samples are randomized (step V) to assay procedure and order of handling (location of plate or thermocycler). The operator is blinded to the concentration (step VI) of each sample.
For option 1, 3 dilutions are created, corresponding to low, medium, and high concentrations (Fig. 2, step I). The material is divided at each concentration into 6 aliquots (Fig. 2, step II), each aliquot having sufficient volume for 10 extractions (and associated reactions), with 5 extractions used for the candidate method and 5 extractions used for the current method. All aliquots are stored (Fig. 2, step III) in an identical manner prior to use to assure similar handling of material (e.g., the same number of freeze–thaw cycles). Aliquots are randomized to the run in which they will be tested.
At the time of testing, the appropriate aliquot is thawed for each of the 3 concentrations, and 10 replicates are created for each concentration, with 5 replicates used for each extraction method (Fig. 2, step IV). At this point, replicates are randomized to extraction method and location on the plate or in the thermocycler. The operator is blinded to the concentration (Fig. 2, steps V and VI).
For option 2, pooled extracted nucleic acid is used to assess modifications affecting only the amplification process. Three dilutions are created consisting of low, medium, and high concentrations of extracted nucleic acid where the low concentration contains sufficient nucleic acid to produce a Ct value for every replicate tested, the high concentration is based on the availability of material and consistent detection by PCR, and the medium concentration produces a response between the responses generated by the high and low concentrations. The extracted material is divided at each concentration into 12 aliquots where each aliquot has sufficient volume to test 5 replicate reactions and is randomized to the assay procedure and testing run. At the time of testing, 2 aliquots of each of 3 concentrations are thawed and further divided into 5 replicates. In total, 15 samples are tested using each method (current and candidate) for each testing run incorporating randomization and blinding.
A single operator conducts all testing to avoid confounding the difference between operators with the assay procedure (candidate vs. current). While the ultimate goal is to evaluate the variability within and between assays, it is impossible to remove the ability of the operator from the experiment. The assumption is that the observed estimates may change if another operator conducted the testing, but the relationship between the candidate and current method remains the same.
Case study—repeatability
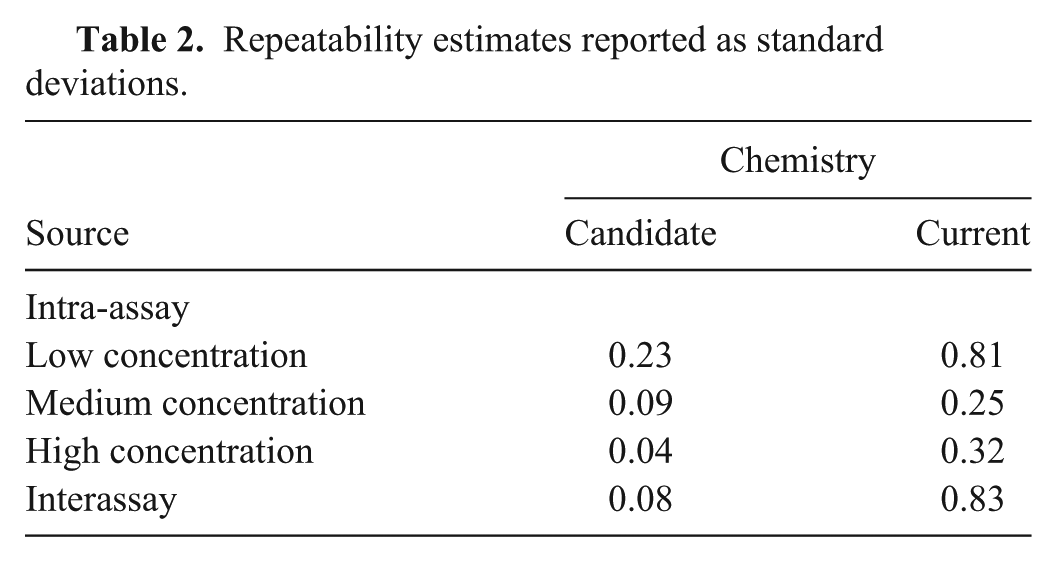
The repeatability experiment used 3 concentrations of the same A22 Iraq isolate used in the LOD experiment. Individual aliquots (15 μl) of extracted RNA for each concentration were tested as 5 replicates for each chemistry (candidate and current) repeated on 6 separate occasions (days). The data is displayed in Figure 3. There was evidence of nonconstant variance between the replicates among concentrations, so an estimate of intra-assay variability is provided for each concentration. Table 2 provides the estimates for inter- and intra-assay variability expressed as standard deviations. The intra-assay standard deviations were lower for the candidate chemistry compared with the current chemistry for all concentrations, and the interassay standard deviation was lower as well.
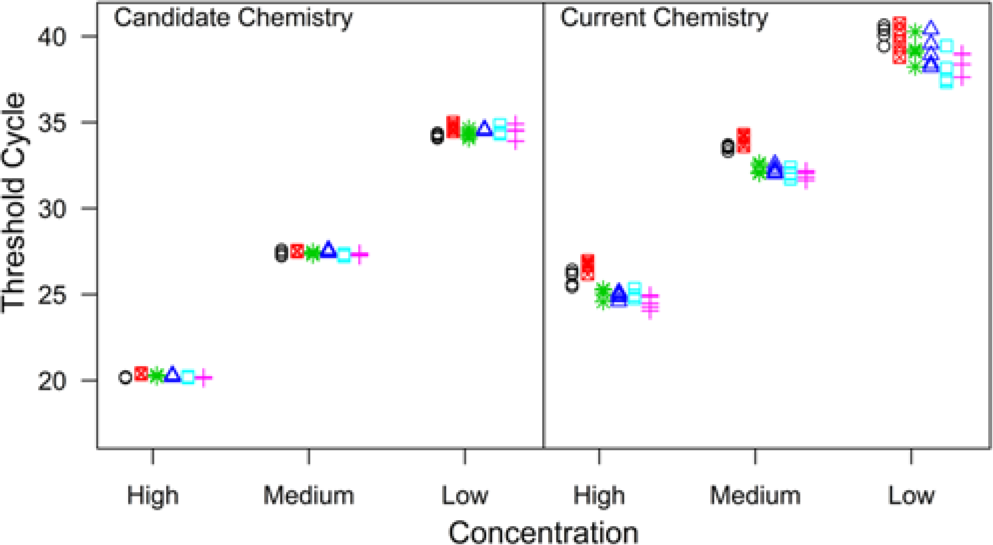
Repeatability data where each plot displays threshold cycle versus concentration of a Foot-and-mouth disease virus A22 Iraq isolate. The left plot is the repeatability data using the candidate chemistry and the right plot is the repeatability data using the current chemistry.
Repeatability estimates reported as standard deviations.
Cross-contamination
Introduction of robotics to replace a manual pipetting process may necessitate evaluation of cross-contamination for the candidate (robotic) method. This experiment is required only for the candidate (robotic) method only. Assuming a 96-well format, highly concentrated positive samples are surrounded on all sides with negative samples, and testing is performed. The operator watches for possible contamination in wells without added template. For the current case study, no cross-contamination study was performed.
Operator variability
An experimental assessment of operator variability is necessary if there is reason to believe that skill may have an impact on the relationship between the candidate and current method. Operator variability is assessed by the testing of 10 independent positive samples spanning the assay’s range and 1 or more negative samples. Again, operators conducting the testing are blinded to the sample status (strong positive, weak positive, negative). The samples are randomized in regard to the order of handling and location on the plate or within the detection platform.
This data does not require a formal statistical analysis. Plots of the raw data will reveal if 1 or more operators are obtaining results inconsistent with the remaining operators. A plot of the Ct value against operator is used where different colors or plotting characters (symbols) distinguish the panel members. Alternatively, a plot of the Ct value against panel member is used where different colors or symbols distinguish operators. Summary information regarding the number of correct classifications for each operator is provided if operator variability is tested.
Stage II testing
Stage II of the validation process for an assay involves estimating diagnostic sensitivity and specificity. 9 Diagnostic sensitivity is the probability of correctly classifying a truly positive specimen, and diagnostic specificity is the probability of correctly classifying a truly negative specimen. In a de novo validation, estimation of diagnostic sensitivity and specificity requires testing many known positive and negative samples, but diagnostic sensitivity and specificity is not reestimated for a minor change. Rather, the NAHLN MTWG uses a minimum of 30 positive and 30 negative diagnostic samples to assess if the candidate method results in an assay that remains fit for its original intended purpose. All diagnostic samples are tested using both the candidate and current method.
Testing diagnostic samples
The goal of this experiment is to determine if diagnostic samples behave in a similar fashion in the 2 test methods. The first step is to identify the source and acquisition of samples. 4 Each sample should originate from a different animal. Serially drawn samples taken on different days from the same animal should be avoided as these are not representative of individual animals in a population. 9 If the performance characteristics of the assay are known to be substantially different based on sample type or animal species, then the samples should be tested from all species and sample types. When warranted, a single analysis is performed combining results across species and/or sample types.
Operators conducting the testing are blinded to the status of each sample, and samples are randomized to position in the instrument. If more than 1 run is required to accommodate the positive and negative samples then a randomization scheme should be used to prevent all negative samples being tested in a single session and all positive samples being tested in a separate session.
Agreement is assessed in multiple ways. First, each of the samples providing a Ct value (not including Ct values reported as 0 or “undetermined”) as tested by both methods are used to estimate the correlation and concordance correlation.5,6 The agreement based on the positive and negative classification is assessed by estimating kappa. 1 The mean difference (candidate minus current) in Ct values are then estimated. Collectively, these estimates allow for the assessment of the diagnostic performance of the candidate method as compared to the current method.
Case study—testing diagnostic samples
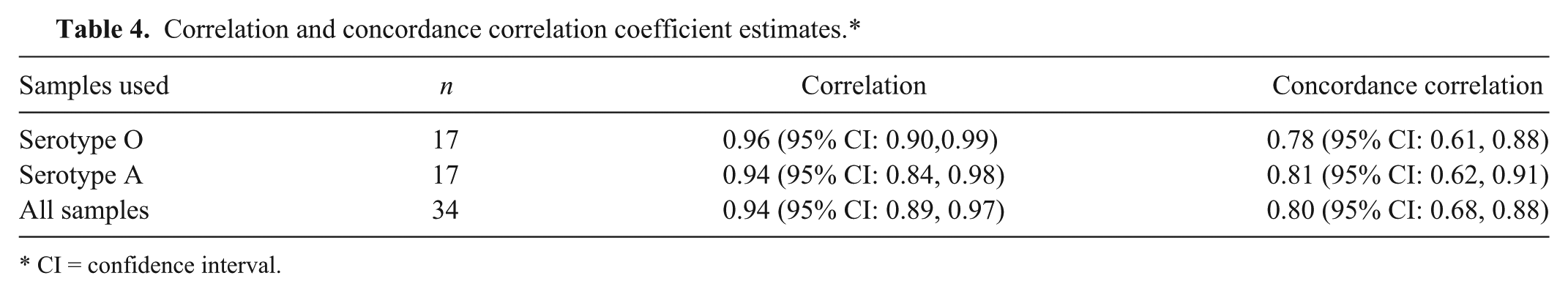
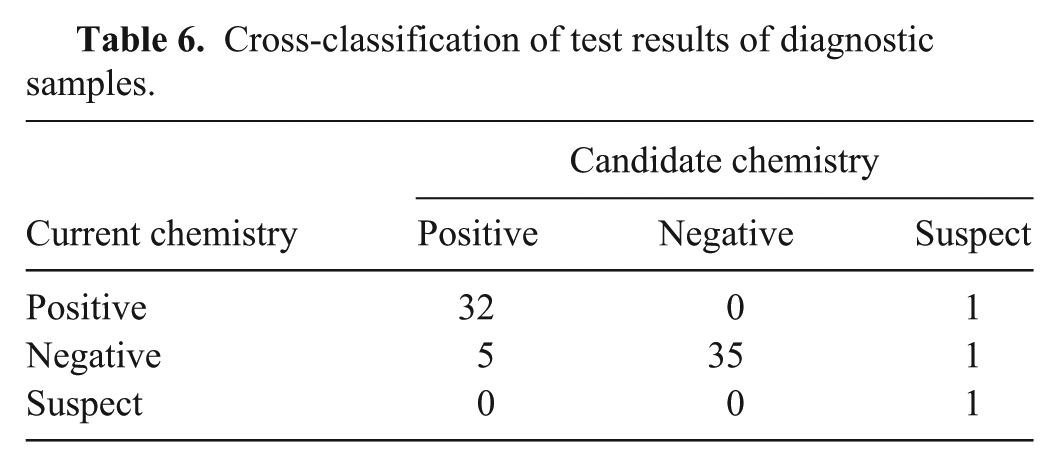
A total of 35 negative and 40 positive diagnostic samples were tested using each chemistry. Among the negative samples tested, 20 were bovine tongue epithelium tissues and 15 were swine tongue epithelium tissues. Each sample was extracted using commercial reagents. Extracted RNA was aliquoted (4 μl per aliquot) for single use. The positive sample set, representing FMDV serotypes O and A, was composed of oral swabs, epithelium, and probang samples from bovine, swine, ovine, and caprine (Table 3). Data was combined in the display (Fig. 4) and analyses. Correlation and concordance correlation were estimated by serotype and by combining all samples (Table 4). The mean difference and associated 95% confidence intervals (CI) are presented in Table 5. The cross-tabulated results are presented in Table 6. Kappa was estimated to be 0.84 (95% CI: 0.72, 0.96) regardless of whether the suspect samples were treated as negatives or as positives.
Number of positive diagnostic sample types by species.

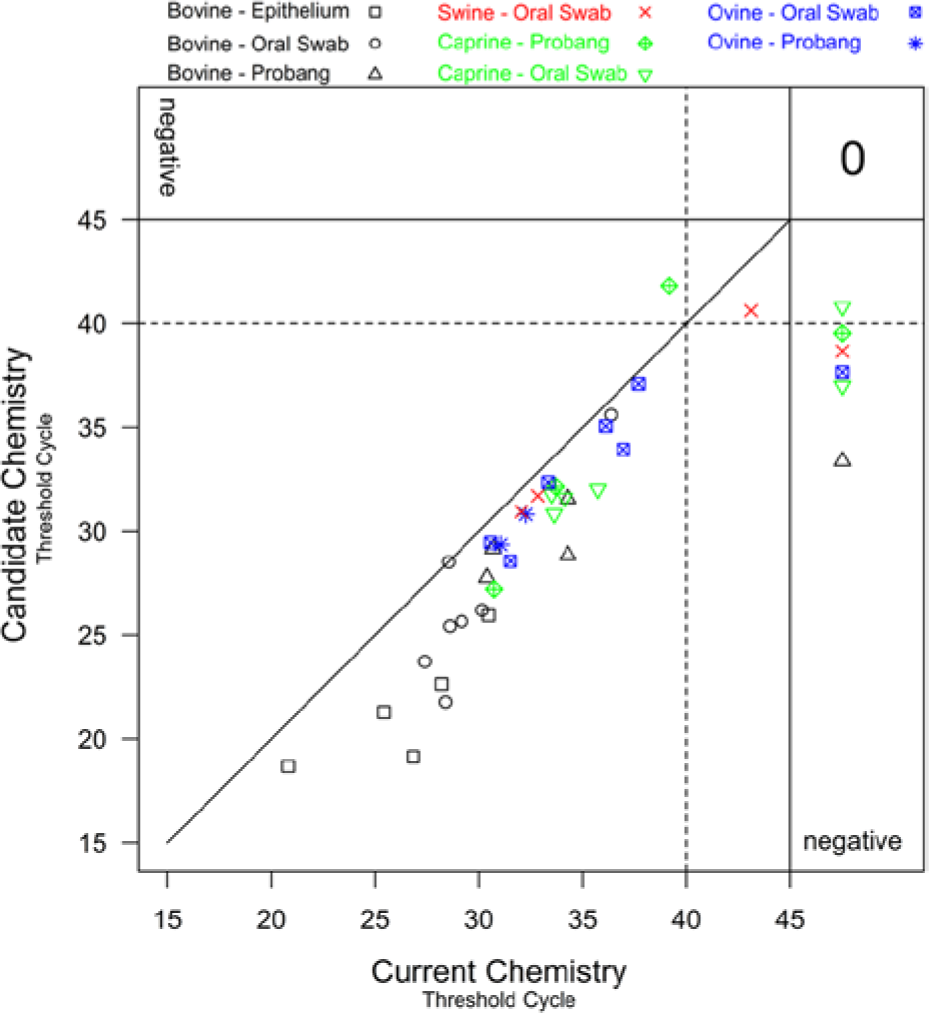
A plot of the threshold cycle obtained using the candidate chemistry against the threshold cycle obtained using the current chemistry for positive diagnostic samples. The different colors and plotting characters represent different species and sample types. The “0” in the upper right hand corner indicates that there were no positive diagnostic samples that tested negative using both the candidate and current method.
Correlation and concordance correlation coefficient estimates.*
CI = confidence interval.
Mean difference (candidate minus current) estimates.*
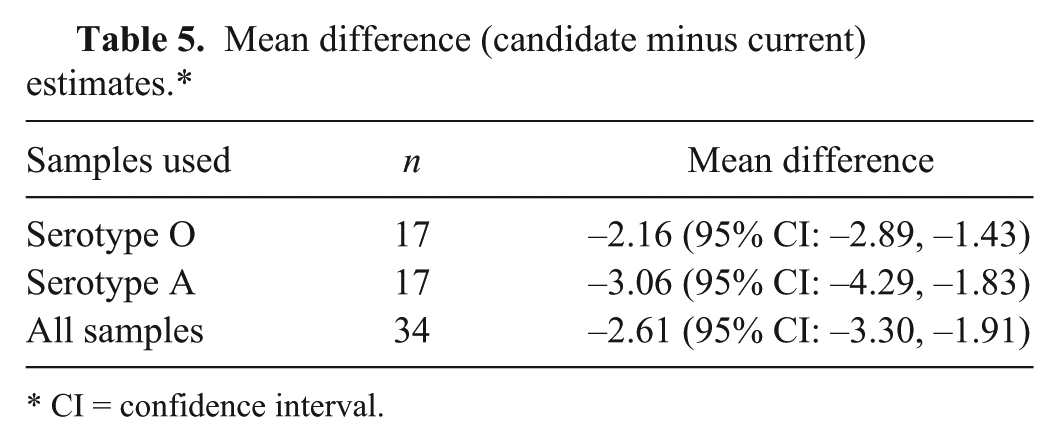
CI = confidence interval.
Cross-classification of test results of diagnostic samples.
Discussion
The current study details the experimental design used by the NAHLN MTWG to assess the impact of a minor change to a validated assay. Experimental design is the primary focus to facilitate proper statistical analysis. To this end, the present review provides detailed descriptions of experimental designs used as a cost effective way to compare a minimally modified assay with a validated assay deployed in the NAHLN.
Throughout the review, explicit details regarding study design are provided to facilitate analysis and limit the need to consider alternate explanations for observed phenomenon. For example, the LOD experiment is conducted on 3 independent plates rather than on a single plate. Note the comparison of the candidate chemistry to the current chemistry using the O Rey isolate (Fig. 1; Table 1). Given a concentration, the Ct values observed for the current chemistry were consistently higher than for the the candidate chemistry. At a concentration of approximately 8 log10 TCID50/ml (Fig. 1), the Ct values observed using the O Rey isolate were approximately 25 and 15 for the current and candidate chemistries, respectively. This corresponds to a difference of 10 Ct units given the concentration. Had the experiment been conducted on a single day on a single plate, the question would remain whether the higher Ct values for the current chemistry were simply due to conditions encountered during a single testing occasion. Because similar results were observed on 3 independent runs using the same aliquots by both the candidate and current chemistries, it is reasonable to conclude that the observed phenomenon is a function of the chemistries and not a technical error in the pipetting of the master mix, in extraction, or differences between operators. The manner in which the testing was conducted eliminates the need for speculation regarding an unintentional laboratory error as the phenomenon was repeatable.
The use of probit regression to estimate the 95% endpoint, commonly used for the LOD, has been discussed previously. 2 Response probabilities between 0 and 100% must be represented to use probit regression. Smaller dilutions and more replicates per dilution likely will be necessary to use a probit regression model to estimate the 95% endpoint. This would require increasing the cost of testing associated with use of additional resources, and such experimentation is not deemed necessary by the NAHLN MTWG for a methods comparison study. Furthermore, probit regression (generalized linear model with a probit link function) assumes the response pattern (percent positive) is symmetric with respect to log10 concentration. If the distribution of genomic copies is believed to be Poisson, a complementary log-log link function would be most appropriate. 7
Variability among the replicates for a given run of the assay was not constant across all concentrations tested. Increasing variance with decreasing concentration was observed in the experiment that compared the candidate and current chemistries (Fig. 3). Visually, it is not possible to separate the variability within the run and the variability among the runs. However, it is possible to use visual indications to obtain a sense of the variability between the runs. Here, it is desired to visually assess the spread between the different plotting symbols given the concentration. For instance, within the high concentration using the current chemistry, observations from the first 2 runs were generally higher than those from the remaining 4 runs and do not fall in an apparent straight horizontal line as when the candidate chemistry was used. The estimates in Table 2 for interassay variability confirm what was observed visually. There was less between-run variability when the candidate chemistry was used.
Pearson correlation coefficient alone is not a measure of agreement. 2 However, the estimation of the concordance correlation coefficient5,6 together with the Pearson correlation, the mean difference, kappa, and observing a plot of the data collectively allows for the assessment of the agreement between the 2 methods.
Estimates for concordance correlation coefficients were notably smaller than those for the correlation coefficient. This is an indication that the observed Ct values will not fall on the 45º line of agreement (Fig. 4). The Ct values produced using the candidate chemistry were generally lower (better) than those produced by the current chemistry. There were several samples in which a Ct value was observed using the candidate chemistry, and no Ct value was observed when using the current chemistry. There was no consistency in terms of species or sample type of the samples that were deemed positive by the candidate method and negative by the current method. The zero in the top right corner of Figure 4 describes the number of positive samples producing negative results by both chemistries. All negative samples tested negative by both methods.
Conclusion
Details have been provided for the design of studies used by the NAHLN MTWG to compare a validated diagnostic test method with a similar method incorporating some minor modification. The results of these experiments have allowed for the evaluation of the minor modification and whether the candidate assay remains fit for its intended purpose.
The LOD estimates for the candidate method were the same or lower (better) than the current method. The amplification efficiency estimates for the candidate method were the same or higher (better) than the current method. The repeatability estimates were consistently lower (better) using the candidate method as compared to the current method. All negative samples tested negative by both methods suggesting that the candidate method was similar to the current method. The candidate method was detecting virus in positive samples that the current method was not. The candidate method consistently produced lower Ct values when testing positive diagnostic samples than the current method. The collective results suggest the candidate method may be superior to the current method. The candidate method was deemed fit for the intended purpose and was accepted as a viable substitution for the current method.
Footnotes
Acknowledgements
The authors would like to thank all members of the National Animal Health Laboratory Network’s Methods Technical Working Group for the valuable advice and contributions to the development of the methods comparison process discussed herein.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the U.S. Department of Agriculture (USDA), APHIS National Veterinary Services Laboratories as part of the animal disease diagnostic mission and by interagency agreement with the USDA Agriculture Research Service under award number 60-1940-7-011.