
Abstract
Brucellosis has emerged as a disease of concern in marine mammals in the last 2 decades. Molecular detection techniques have the potential to address limitations of other methods for detecting infection with Brucella in these species. Presented herein is a real-time polymerase chain reaction (PCR) method targeting the Brucella genus–specific bcsp31 gene. The method also includes a target to a conserved region of the eukaryotic mitochondrial 16S ribosomal RNA gene to assess suitability of extracted DNA and a plasmid-based internal control to detect failure of PCR due to inhibition. This method was optimized and validated to detect Brucella spp. in multiple sample matrices, including fresh or frozen tissue, blood, and feces. The analytical limit of detection was low, with 95% amplification at 24 fg, or an estimated 7 bacterial genomic copies. When Brucella spp. were experimentally added to tissue or fecal homogenates, the assay detected an estimated 1–5 bacteria/µl. An experiment simulating tissue autolysis showed relative persistence of bacterial DNA compared to host mitochondrial DNA. When used to screen 1,658 field-collected marine mammal tissues in comparison to microbial culture, diagnostic sensitivity and specificity were 70.4% and 98.3%, respectively. In addition to amplification in fresh and frozen tissues, Brucella spp. were detected in feces and formalin-fixed, paraffin-embedded tissues from culture-positive animals. Results indicate the utility of this real-time PCR for the detection of Brucella spp. in marine species, which may have applications in surveillance or epidemiologic investigations.
Introduction
Distinct marine mammal-associated strains of Brucella were first isolated in 1994 from both Atlantic and Pacific Ocean species.10,29 In subsequent years, these pathogenic bacteria have been found to have worldwide distribution, with exposure to, or infection with, Brucella spp. documented in many cetacean, pinniped, and marine carnivore species.14,26,27 Brucella pinnipedialis and Brucella ceti have been proposed as formal names for isolates from pinnipeds and cetaceans, respectively, based on metabolic differences in culture and genetic analyses. 11 Other genetic studies support 3 distinct lineages with associated host preferences for pinnipeds, dolphins, and porpoises, although host specificity is not absolute. 13 Information about exposure and disease prevalence in potentially vulnerable marine mammal populations is growing; however, many questions have yet to be answered, including the specifics of transmission, pathogenicity, and susceptibility.
Numerous serologic methods have been used to assess exposure to Brucella, with variable rates of seropositivity reported in a number of species of North Atlantic and Pacific marine mammals, suggestive of enzootic exposure in some populations.19,26,30 Clinical manifestations of brucellosis in marine mammals are so far described only in cetaceans, with abortion and placentitis, orchitis, visceral and blubber abscesses, musculoskeletal lesions, and meningoencephalitis reported.12,20,27 High levels of seropositivity are frequently reported in pinnipeds, and microbiologic isolation is reported in many pinniped species. To date, however, no definitive evidence of associated pathologic change has been described in this taxonomic group, suggesting that the presence or absence of Brucella-specific antibodies alone may not adequately assess infection status in individuals or populations of pinnipeds.26,31 Inconsistent seroconversion with known infection has been observed. 18 Clinical interpretation of positive titers in stranded pinnipeds during rehabilitation or in captive populations is obfuscated by the absence of detectable disease.
Microbiologic culture is considered the “gold standard” for diagnosis of brucellosis, but drawbacks include availability of suitable premortem tissue samples, requirement of specialized media, long incubation times, and the potential for exposure of laboratory personnel to this zoonotic agent.4,6,7 Poor postmortem carcass condition and prolonged storage of tissues from marine mammal species may also be hypothesized to reduce the success of attempts to isolate these fastidious organisms. Focal infections may also not avail themselves to standard isolation techniques. 23
Molecular techniques, including conventional and real-time polymerase chain reaction (PCR) assays, are being used with greater frequency for species identification of Brucella culture isolates, as well as for diagnostic testing of clinical samples. 7 In human diagnostics, PCR techniques have been found to be more sensitive than serologic tests and are considered useful for confirmation of acute or relapsing illness due to Brucella in cases where serology or culture have been nondiagnostic.9,21 Veterinary diagnostic applications of PCR have been developed for blood-, milk-, and tissue-based detection of Brucella spp. in domestic species.16,17
Published studies where PCR has been used to survey for Brucella in marine mammal tissues, with or without microbiological culture, are uncommon. 27 Molecular detection techniques have the potential to address shortcomings associated with other methods of diagnostic testing for Brucella spp. in marine mammals, including questions of sample access, quality and quantity, and limited availability of host species–specific antibody detection reagents. Presented herein is a real-time PCR method developed for rapid screening of marine mammal tissues. The PCR targets the Brucella genus–specific bcsp31 gene and includes 2 quality control targets to assess the suitability of extracted DNA for amplification and the presence of inhibitors of PCR.
Materials and methods
Brucella spp. and other bacterial controls
Positive controls for PCR were methanol-killed cultures of Brucella pinnipedialis from a harbor seal (Phoca vitulina) a and Brucella ceti from a bottlenose dolphin (Tursiops truncatus). b Brucella abortus standard tube and complement fixation antigen, c a whole cell bacterial lysate, was used as a source of DNA from a nonmarine Brucella strain. A small set of 6 nontarget bacteria—Staphylococcus aureus (American Type Culture Collection [ATCC] 25923), Pseudomonas aeruginosa (ATCC 27853), Escherichia coli (ATCC 25922), and field isolates of Aeromonas hydrophila, beta-hemolytic Streptococcus sp., and Vibrio sp.—were supplied by a veterinary microbiology laboratory. d Concentrations of suspensions of killed Brucella were estimated by ultraviolet spectroscopy e using a conversion factor of 0.15 optical density at 600 nm/109 cells/ml (M. Roop, unpublished data, 2005). DNA was extracted from aliquots of bacterial suspensions using the protocol for Gram-negative bacteria of a commercial extraction kit. f Non–Brucella bacteria were subjected to an additional lysozyme digestion (0.1 mg/ml) at 37°C followed by incubation at 95°C for 10 min before addition of lysis buffer. DNA extracts were quantified by ultraviolet spectroscopy by measurement of absorbance at 260 nm. Copy number calculations assumed a 3.3-Mb Brucella genome, with a calculated genomic molecular weight of 2.14 × 1010 g/mol (0.28 genomic copies/fg).
Extraction of DNA from fresh and paraffin-embedded tissues, blood, and feces
Marine mammal tissues were collected and submitted by collaborators and accepted under National Marine Fisheries Service permit no. 42-1642, and procedures were approved by the Mystic Aquarium & Institute for Exploration’s Institutional Animal Care and Use Committee. Species included Arctocephalus townsendi, Balaena mysticetus, Balaenoptera acutorostrata, Balaenoptera physalus, Callorhinus ursinus, Cystophora cristata, Delphinapterus leucas, Delphinus delphis, Enhydra lutris kenyoni, Enhydra lutris lutris, Enhydra lutris nereis, Erignathus barbatus, Eschrichtius robustus, Eubalaena glacialis, Eumetopias jubatus, Globicephala melas, Grampus griseus, Halichoerus grypus, Kogia breviceps, Lagenorhynchus acutus, Lontra canadensis, Megaptera novaeangliae, Mesoplodon densirostris, Mirounga angustirostris, Orcinus orca, Phoca groenlandica, Phoca vitulina, Phocoena phocoena, Phocoenoides dalli, Stenella attenuata, Stenella coeruleoalba, Tursiops gilli, Tursiops truncatus, Zalophus californianus, and Ziphius cavirostris. Tissues were collected by collaborators at necropsy and shipped frozen; after receipt, tissues were frozen at −80ºC until use. Tissues included adrenal gland, blood and blood products, blubber, bone, brain, colon, epididymis, feces, kidney, liver, lung, lymph node, mammary gland, ovary, placenta, skin, spleen, testis, thymus, thyroid, tonsil, and uterus. Total DNA was extracted from tissues using a commercial kit, f with overnight incubation in lysis buffer and proteinase K at 55°C, and final elution in 100 µl of elution buffer. Scrolls of formalin-fixed, paraffin-embedded tissue (40 µm thick) were deparaffinized by 3 rounds of washing in 1 ml of xylene followed by centrifugation, then 3 rounds of washing in 1 ml of absolute ethanol followed by centrifugation, before extraction as previously described. Blood samples were taken from a seronegative captive beluga whale (Delphinapterus leucas) by standard phlebotomy techniques during routine veterinary examinations 5 and collected into potassium–ethylenediamine tetra-acetic acid (EDTA) blood tubes. g Whole blood and separated blood components were extracted according to a supplemental protocol for purification of total DNA from animal blood. f Fecal samples were extracted using a commercial kit. h
Brucella primers and probe
Previously described primers and probe for the bcsp31 gene target were used for genus-specific detection of Brucella sp. bacteria 28 (bcsp31 forward: 5’-GCTCGGTTGCCAATAT CAATGC-3’; bcsp31 reverse: 5’-GGGTAAAGCGTCGC CAGAAG-3’; bcsp31 probe: 5’-FAM-AAATCTTCCA CCTTGCCCTTGCCATCA-BHQ1-3’). All primers and probes were commercially synthesized. i
Design and construction of internal control target
An amplification control plasmid was created with an inserted sequence of hybrid DNA, which included bcsp31 primer sites and an internal sequence from Atlantic salmon (Salmo salar) beta-globin gene (GenBank accession no. X97287). Primer and probe sites were chosen from the salmon gene with melting temperature and length similar to the interprimer bcsp31 gene sequence. j Genomic DNA obtained from Atlantic salmon blood was amplified by conventional PCR using hybrid primers including sequences of the bcsp31 forward or reverse primers added to the 5’-terminus of respective salmon beta-globin gene primer sequences (hybrid forward: 5’-GCTCGGTTGCCAATATCAAT GCCGTGTGTGGAGCTCTGGATA-3’; hybrid reverse: 5’-GGG TAAAGCGTCGCCAGAAGTTTCAGGGTCGACG AAGAGT-3’ [italics denote S. salar beta-globin genetic sequence]). Total reaction volume was 50 µl including DNA template (335 ng), 200 nM primers, 200 µM of each deoxynucleoside triphosphate, 1 IU Taq polymerase, k and 5 µl of 10 × PCR buffer. k Amplification began with a 10-min activation step at 95ºC, followed by 35 cycles of denaturation at 95ºC for 1 min, annealing at 55ºC for 1 min, and elongation at 72ºC for 1 min, followed by a final elongation for 10 min at 72ºC. The resulting gel-purified, A-tailed PCR product was cloned into a commercially sourced plasmid.l,m,n The 149-bp hybrid plasmid insert was confirmed by PCR amplification using the bcsp31 primers (5’-GCTCGGTTGCCAATATCAATGCCGTGTGTGGAGCTCTGGATAAAGCTGTGAAGAACATGGGCAACATCTTGGCCACATACAAGTCACTGAGCGAGACCCACGCCAACAAACTCTTCGTCGACCCTGAAACTTCTGGCGACGCTT TACCC-3’) and sequencing. i Detection of this plasmid in the multiplex reaction used bcsp31 gene primers and a probe targeting the plasmid insert (beta-globin probe: 5’-JOE-TGGCC ACATACAAGTCACTGAGCGA-BHQ1-3’).
Design of DNA quality-control target
To address concerns about the quality of extracted DNA, a mitochondrial gene target for the marine mammal hosts was included. Fifteen mitochondrial 16S ribosomal RNA gene sequences from pinniped and cetacean species (Table 1) were aligned, and a 216-bp conserved sequence that was conserved across eukaryoteso,p,q was identified. Forward and reverse primers and an internal probe site were identified within this sequence, j with melting temperatures and length characteristics similar to those of bcsp31 gene primers and probe (Mito forward: 5’-CTAGGGATAACAGCGC AATC-3’; Mito reverse: 5’-GGTCTGAACTCAGATCACG TAG-3’; Mito probe: 5’-ROX-ACGACCTCGATGTTGG ATCAGGACA-BHQ2-3’).
16S mitochondrial gene sequences used for creation of DNA quality-control target.

Multiplex real-time PCR conditions
Reaction conditions and primer and control plasmid concentrations were optimized for detection of a low copy-number Brucella infection together with reliable amplification of both internal controls. All PCR reactions were performed in 20-µl total volume in 96-well plates. r Each reaction mixture contained 0.6 μm bcsp31 probe, 0.3 μm bcsp31 primers, 0.2 μm mitochondrial 16S primers, 0.2 μm mitochondrial 16S and beta-globin probes, 1 fg control plasmid (when included), 6.5 mM MgCl2, and PCR mastermix. s Template sample volume varied from 1 to 4 µl, with RNase- and DNase-free water used to adjust the final reaction volume. Amplification began with an initial 95°C activation step for 15 min, then 45 cycles of 95ºC denaturation for 30 sec and 63°C annealing/extension for 60 sec. Fluorescence data was collected at the end of the annealing/extension step.
Target specificity testing, assay variability, and analytical limit of detection
DNA extracts of B. pinnipedialis, B. ceti, and B. abortus (positive controls), nontarget bacterial species, and marine mammal host DNA were tested using all 3 components of the assay, singly and in multiplex conditions. Four independent assays were performed on 4 different days, 2 each by 2 technicians. Serial dilutions were made of DNA from B. pinnipedialis; 4 µl of each dilution were used as template, with final concentrations of 500 fg, 100 fg, 50 fg, 10 fg, and 5 fg bacterial DNA per reaction. To test for amplification competition, 3 combinations of template DNA were compared: bacterial DNA only, bacterial DNA and internal control plasmid (1 fg), and bacterial DNA, internal control plasmid (1 fg), and harbor seal genomic DNA (100 ng).
Baseline fluorescence data for each real-time PCR assay were manually adjusted to values averaged between 15 and 27 cycles. A fluorescence threshold was chosen within the exponential phase of amplification that maximized the correlation coefficients of the standard curves, and this value was used for all validation runs. Regression lines were compared using linear models and a test for slope heterogeneity. Finding that the slopes of the 3 lines did not differ significantly, analysis of covariance and comparison of least square means were used to compare intercepts. Comparison of percentage amplification was done by χ2 analysis. The limit of detection (LOD; i.e., concentration of bacterial DNA resulting in amplification 95% of the time) was calculated by interpolation of the linear regression of percentage amplification versus log transformed concentration at the 3 lowest concentrations.t,u,v
Sample matrix, tissue degradation, and inhibition studies
To assess the ability of the assay to detect Brucella spp. in a variety of sample matrices, experimental addition of methanol-killed bacteria to tissue, blood, or feces was performed. A homogenate of 1:1 sterile phosphate buffered saline (PBS) w and culture-negative spleen from a harbor seal was prepared by maceration x and spiked with killed B. pinnipedialis. Aliquots of PBS were spiked with the same numbers of bacteria. Aliquots (25 µl) of spleen homogenates or PBS were extracted as previously described. To examine degradation of the mitochondrial target with tissue autolysis, spiked aliquots of homogenates were incubated at room temperature, and samples were taken for extraction at 2, 4, 8, and 16 days after preparation. Freshly drawn whole blood was spiked with killed harbor seal–origin Brucella bacteria and incubated while rocking at room temperature for 1 hr. Aliquots of 50 μl of whole blood were collected, and the remaining blood was centrifuged to separate plasma, buffy coat, and packed red blood cells to examine possible sample partitioning. Total DNA was extracted separately from 50 μl of each blood component. Feces from a seronegative wild-caught harp seal (Phoca groenlandica) were diluted 1:1 with sterile PBS and spiked with killed bacteria, as previously described. DNA was extracted from 100-μl fecal aliquots, including a negative control.
To evaluate the function of the assay’s plasmid-based inhibition control, substances inhibitory to PCR were added to reactions also containing 1 fg of plasmid, 100 ng of harbor seal splenic DNA extract, and 50 fg of B. pinnipedialis DNA. To simulate incomplete removal of anticoagulants (potassium EDTA and sodium heparin), solutions made from blood collection tubes g filled to volume with water were added, representing final concentrations of 0.27 M EDTA and 5.7 U/ml heparin. Additional harbor seal genomic DNA (100, 200, and 300 ng) was added to other wells.
PCR and culture of field samples
For bacterial culture, approximately 5-g tissue samples were thawed, dipped in absolute ethanol, and flame sterilized externally, then macerated with equal volumes of sterile 0.9% buffered saline. The resulting homogenates were inoculated onto trypticase soy agar plates containing 5% bovine serum, 7.5 U/ml bacitracin, 30 µg/ml cycloheximide, and 1.8 U/ml polymyxin B. 3 Cultures were incubated at 37°C in 10% carbon dioxide for a minimum of 3 weeks. Bacterial isolates with colonial morphology consistent with Brucella spp. were submitted to the National Veterinary Services Laboratory (NVSL) for identification. DNA was extracted from 50 µl of homogenates prepared for culture (as previously described) or from 10 to 25 µg tissue subsections. DNA extracts were assayed in duplicate together with negative (no template) and low (5–10 fg), medium (50 fg), and high (100–500 fg) positive amplification controls.
Results
Target specificity
The PCR assay amplified bcsp31 from B. pinnipedialis–, B. ceti–, and B. abortus–positive controls. Six nontarget bacterial DNA extracts did not amplify the bcps31 or mitochondrial targets. No amplification of pinniped or cetacean host genomic DNA was observed for the bcsp31 target or plasmid control. Amplification of the mitochondrial 16S ribosomal RNA target was demonstrated in all mammalian control tissues.
Assay variability and analytical limit of detection
Linear regressions were performed on standard curves, plotting log concentration against threshold cycle (Ct; Fig. 1). Interassay variability data and correlation coefficients are detailed in Table 2. In all 3 reaction types (with and without internal control plasmid or genomic DNA), there was no statistical difference between technicians in slope or intercept from unweighted least squares analyses (P > 0.05 in all cases); data from both technicians was pooled for further analysis. Comparison of reaction type standard curves showed no difference in slope (P = 0.12) but a significant difference in intercept (P < 0.0001). The intercept for reactions including only bacterial DNA was significantly different from each of the other types (P < 0.0001), which were not significantly different from each other (P = 0.077). Percentage amplification for each reaction combination was not significantly different by χ2 analysis (P = 0.98). Limits of detection for each reaction type are shown in Table 2, calculated by interpolation. The LOD for pooled data is 24 fg, or approximately 7 bacterial DNA copies. Amplification of 5 fg (1.4 bacterial DNA copies) was seen in 79% of sample replicates.
Reproducibility of the Brucella polymerase chain reaction assay for 3 types of template DNA.*
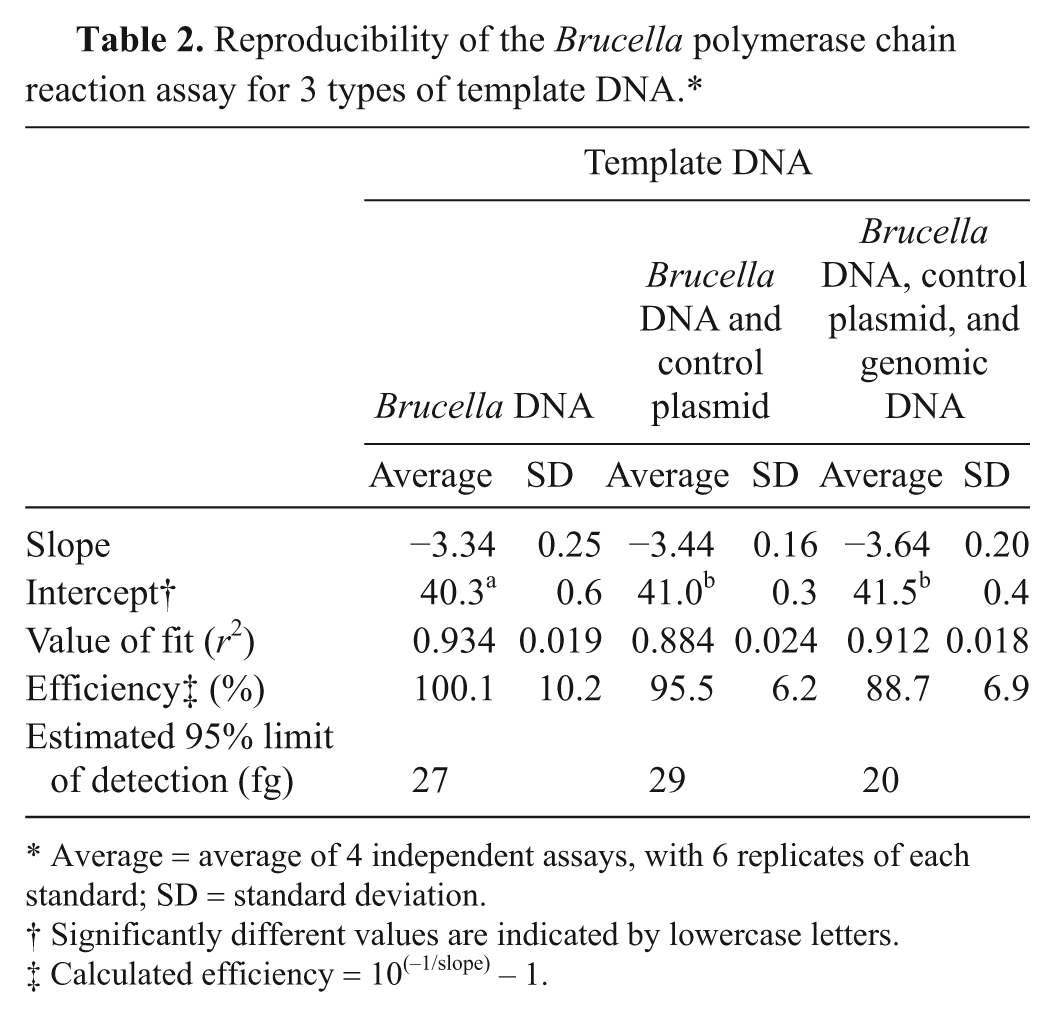
Average = average of 4 independent assays, with 6 replicates of each standard; SD = standard deviation.
Significantly different values are indicated by lowercase letters.
Calculated efficiency = 10(−1/slope) − 1.
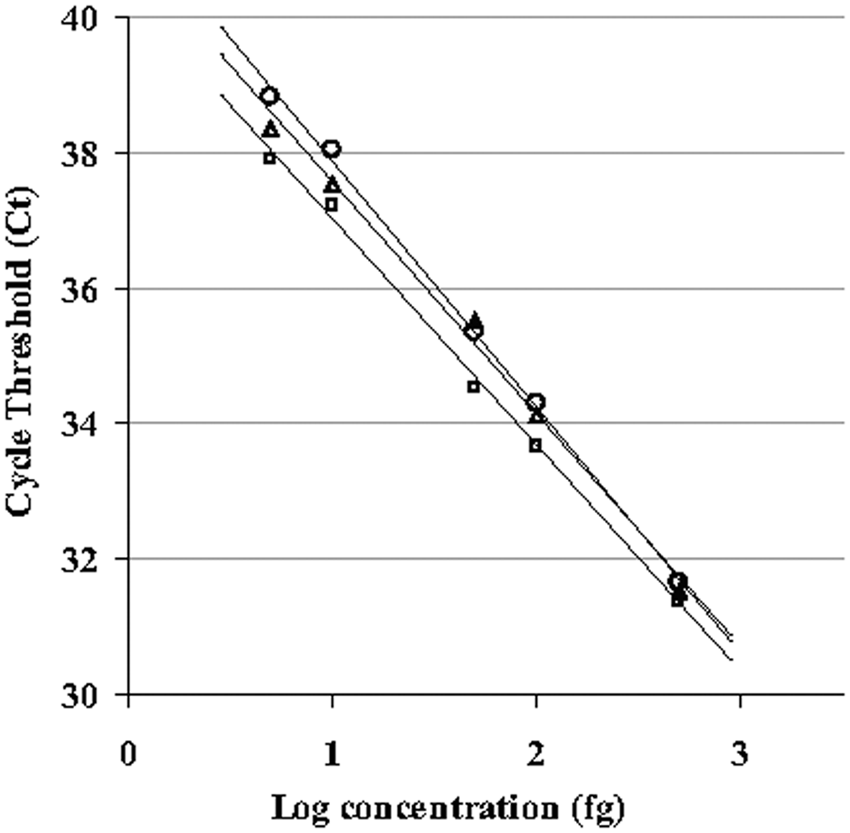
Comparison of real-time polymerase chain reaction standard curves for assays with and without internal control plasmid and harbor seal (Phoca vitulina) genomic DNA. Serial dilutions of a standard DNA extraction of harbor seal–origin Brucella ranging from 5 to 500 fg were used to generate standard curves. Each point is an average of between 19 and 24 independent amplifications. Squares indicate bacterial DNA; triangles indicate bacterial and plasmid DNA; circles indicate bacterial, plasmid, and genomic DNA.
Performance and interpretation of amplification controls
Both mitochondrial 16S and plasmid targets were always positive under normal conditions, with Ct cut-off values of 30 and 40, respectively. If the mitochondrial 16S control target failed to amplify, but the plasmid beta-globin target was positive, the sample was designated as “failed” due to insufficient amplifiable mitochondrial DNA. If the sample amplified the mitochondrial 16S control target but did not amplify the plasmid beta-globin target, the sample was designated as “failed” due to partial inhibition of amplification. Because the plasmid concentration was low, resulting in high Ct values for this target, and mitochondrial DNA was typically abundant, partial but not complete amplification inhibition frequently prevented amplification of the plasmid but allowed amplification of the latter target at a higher Ct. If both controls were positive but the bcsp31 target did not amplify, the sample was considered negative.
Sample matrix, tissue degradation, and inhibition results
All concentrations of Brucella bacteria added to PBS, tissue homogenates, and feces demonstrated positive amplification of the bcsp31 target (Table 3). Only the plasma component of blood amplified the bcsp31 target. Including the extraction process, this testing indicates an effective diagnostic (vs. analytical) LOD of approximately 1 to 5 bacteria/µl of homogenized tissue (2–10 bacteria/PCR reaction). The PCR performed on samples allowed to undergo degradation at room temperature showed significant loss of amplifiable mitochondrial 16S DNA after 8 and 16 days, as indicated by an increase in Ct (Fig. 2). The difference in average Ct between days 0 and 16 was 7.8 cycles, representing a 225-fold effective difference in amplifiable mitochondrial DNA concentration; amplification of the bcsp31 target was demonstrated in every replicate, without degradation over time. Addition of host genomic DNA up to 400 ng was not inhibitory, nor was addition of EDTA (one-way analysis of variance, P = 0.54). Addition of heparin resulted in complete inhibition in all 3 channels.
Results of real-time polymerase chain reaction assay on tissue, feces, and blood spiked with killed marine Brucella.
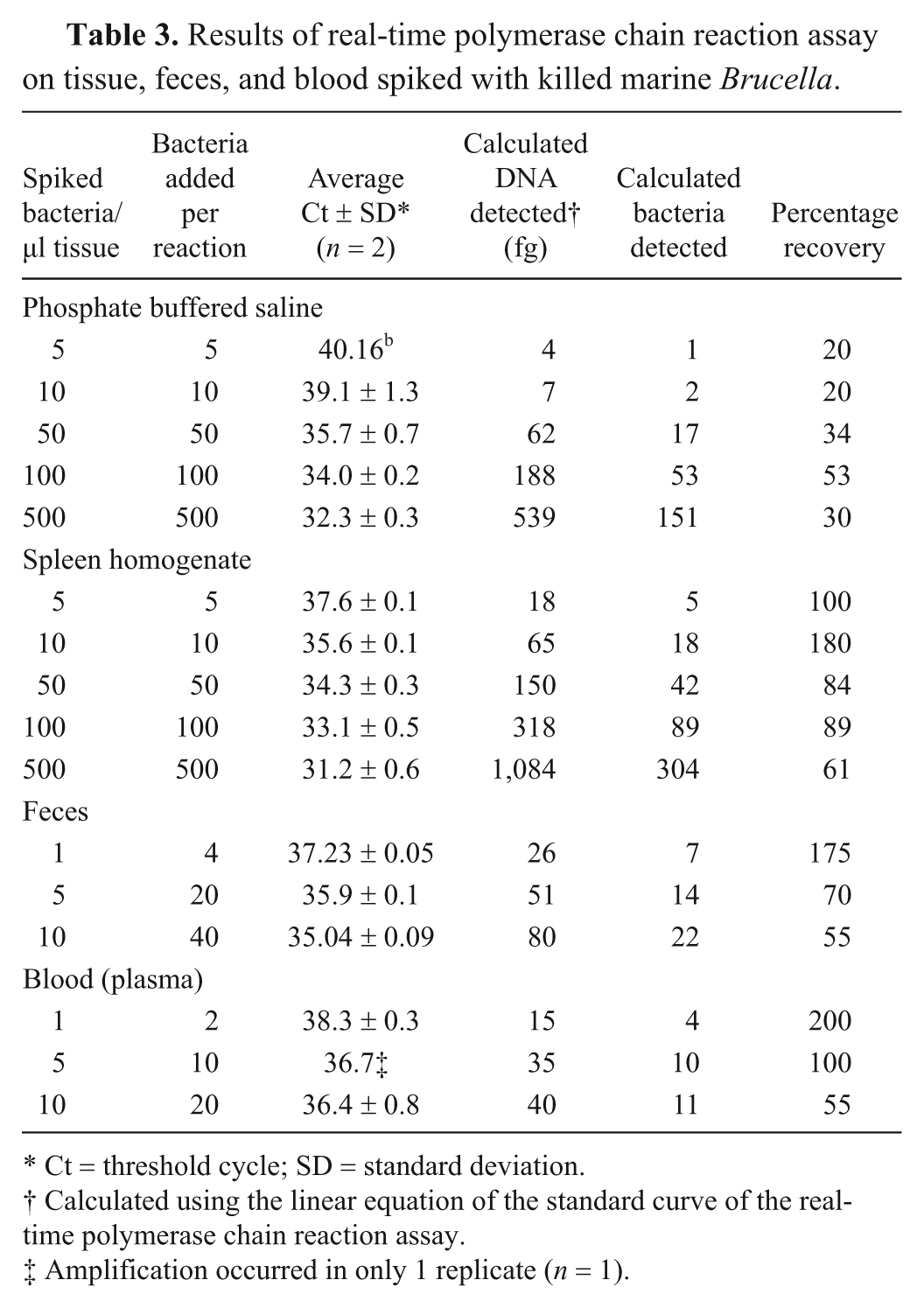
Ct = threshold cycle; SD = standard deviation.
Calculated using the linear equation of the standard curve of the real-time polymerase chain reaction assay.
Amplification occurred in only 1 replicate (n = 1).
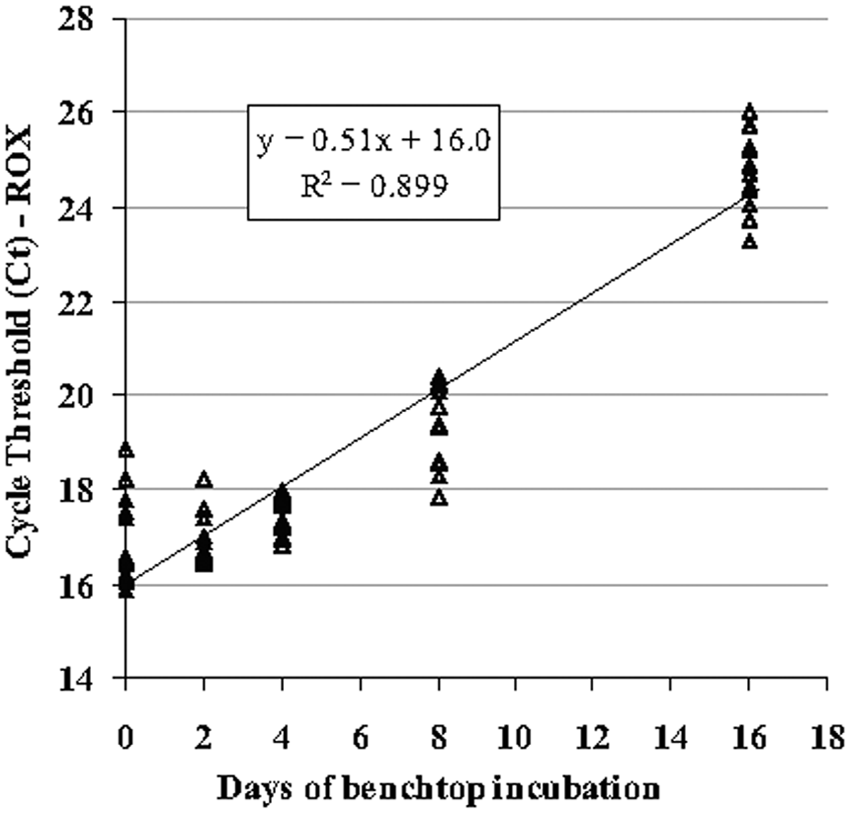
DNA amplification of the mitochondrial target in bacterially spiked spleen homogenates sampled serially over 16 days of autolysis at room temperature. Note loss of amplifiable DNA over the period of degradation (n = 12 at each time point) demonstrable by increasing threshold cycle (Ct) values. ROX = 5-carboxy-X-rhodamine.
Comparison of PCR and culture of field samples
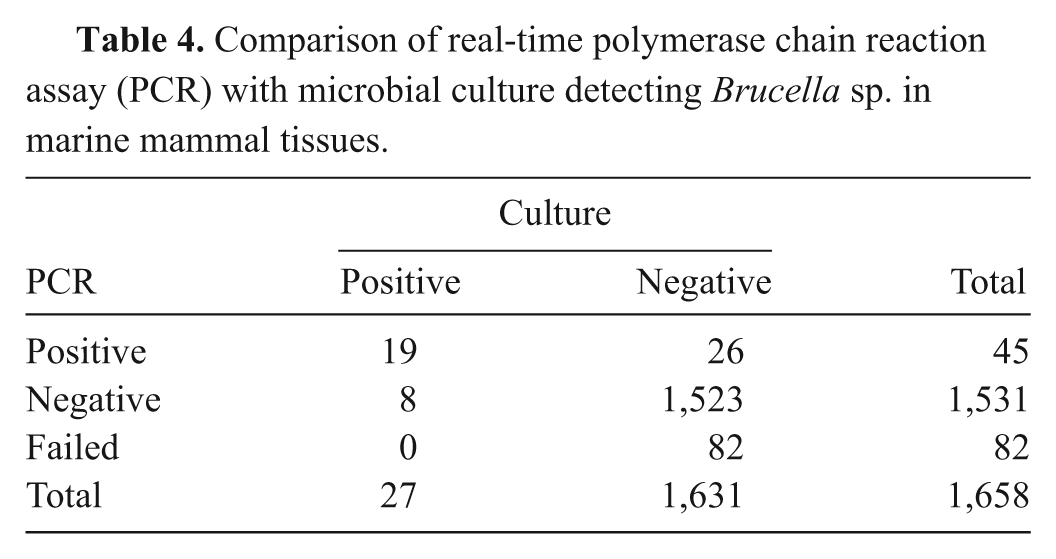
Frozen tissues from 786 individual animals of 35 species of marine mammal were tested by both culture and the described PCR assay, including internal controls. Fifty-three tissues were identified in 32 animals as positive by either or both methods, including blubber, brain, kidney, liver, lymph node, lung, lungworm, placenta, spleen, testis, and tonsil. All positive cultures were confirmed as marine-origin Brucella by NVSL, but were not typed to species. Comparison of culture and PCR testing is summarized in Table 4. All samples failing PCR quality control were failed because of inhibition (absence of plasmid control amplification), not DNA quality (presence of mitochondrial control amplification). The PCR and culture results agreed in 97.8% of cases (both tests were either negative or positive; kappa = 0.52, P < 0.001). Diagnostic sensitivity compared to culture was 70.4%, and specificity was 98.3%. In 26 cases where PCR was positive but culture was negative, another tissue from the same animal (where other samples were available) tested positive by either culture or PCR in 11 of 19 cases. Calculated amounts of DNA detected in these PCR-positive and culture-negative samples (n = 26) averaged 57 fg, or approximately 16 bacteria (±26). Of the 8 samples (6 animals) that were culture-positive but PCR-negative, 2 were PCR-positive in another tested tissue.
Comparison of real-time polymerase chain reaction assay (PCR) with microbial culture detecting Brucella sp. in marine mammal tissues.
Fifty-eight paraffin blocks containing mixed tissues (frequently, more than 1 tissue per block) from 13 animals were tested by PCR, including internal controls. Eleven blocks from 8 animals were positive by PCR. Paraffin block PCR testing for specific tissues agreed with corresponding fresh tissue PCR and/or culture results (where available) in 22 out of 35 (62.9%) of cases. Eight of 9 (88.8%) culture-positive animals were positive by PCR in at least 1 paraffin block. Eight blocks from 4 animals were negative but did not have tissue PCR or culture results available for comparison.
Fecal samples or fecal swabs from 39 animals were tested by PCR. Four were positive, including 3 from animals PCR- or culture-positive in other tissues; in the fourth, other tissues were not available. Six animals positive by PCR or culture in other tissues had negative fecal PCR results. Four failed due to PCR inhibition and were not retested.
Two hundred forty-four blood samples from 227 animals were tested by PCR; all were negative. Fifty-six of these animals had other tissues tested by culture or PCR, 2 of which were positive by these other methods.
Discussion
The objective of the current study was to design a robust but sensitive PCR assay suitable for high-throughput screening of clinical and necropsy samples from marine mammals for the presence of Brucella spp. genomic DNA. The chosen Brucella-specific target, bcsp31, is a conserved single-copy gene coding a 31-kDa outer membrane protein found in all strains of Brucella to date, including marine species. A genus-specific target was chosen to check for all possible species of Brucella in marine animals, for the widest possible utility, with further identification of individual species and strains possible after PCR screening. The described primer-probe combination has been extensively tested for bacterial target specificity against 59 non–Brucella bacteria with no positive amplification. The bcsp31 gene is a commonly used target for detection of Brucella by PCR, and the small size of the amplicon (151 bp) facilitates its use in real-time PCR.1,8,9,29
The developed assay performed well in an analytical sense, detecting very low concentrations of Brucella genomic DNA. The calculated LOD is comparable to, or better than, that reported from other assays developed for the detection of Brucella from clinical samples.2,25,28 Reaction conditions for this assay were optimized for detection of a putatively low copy-number Brucella target against a background of host genomic and plasmid DNA, accounting for competitive binding of primers to both plasmid and Brucella DNA targets. Although standard curve parameters were subtly altered with addition of plasmid and genomic DNA, there was no reduction in percentage amplification at very low bacterial DNA concentrations that would significantly affect LOD.
Previously described inhibitors to PCR include hemoglobin, heparin, EDTA, and excess DNA concentrations, which can result from incomplete purification of DNA extracts or inaccurate DNA quantification.22,24,32 The authenticity of a negative PCR result is supported by knowing amplification is possible using the same bacterial target primers in the same reaction tube. Using this plasmid-based amplification target, complete inhibition due to heparin was detected, but not EDTA. Results of the present study differ from those of a previous study 24 that found decreased sensitivity in PCR reactions with addition of human genomic DNA; however, another study 15 found similar results, suggesting that concentrations of DNA below a certain threshold may not inhibit PCR amplification.
The mitochondrial DNA quality control target was included to detect failure of a reaction due to insufficient or degraded sample, considered likely in samples from wild-stranded marine mammal carcasses that may have undergone significant autolysis. Although the variable number of mitochondria in different organs precludes absolute comparisons of DNA quantity, this target has utility as an indicator of the absolute ability of the sample to support PCR. In the tissue studies described herein, adequate amounts of DNA were detected in samples from whole tissues, swabs, feces, and blood components of low cellularity such as plasma, although smaller amounts were present in extracts from swabs or fluid samples, as compared to solid tissues. Because Ct values for this target were generally low (i.e., host DNA concentrations were high), this target amplified even when inhibition was detected in the other 2 channels, except with addition of heparin to reaction mixtures where total inhibition was seen. In practice, the failure rate of the mitochondrial target was negligible, reflecting pipetting errors rather than tissue degradation or low DNA concentration.
Amplification of host and bacterial DNA was possible even in the face of significant degradation, as was demonstrated in the simulation of postmortem autolysis. Mitochondrial DNA did not show measurable loss until 8 days of incubation, and there was no effective loss of Brucella DNA amplification over the entire 16-day experiment. These results may reflect the relative stability of bacterial DNA compared to that of the host, suggesting that bacterial PCR may be successful even in situations of poor tissue quality. Because the mitochondrial target does not act as a surrogate for bacterial DNA quality, inclusion of this target may not be necessary for that purpose.
Percentage detection of whole cell bacteria varied by tissue, with experimental detection of an estimated 1–5 bacteria/µl in several sample matrices (e.g., tissue, blood, and feces). Because this process includes DNA extraction, differences in extraction efficiency between methods may affect recovery and detection of bacteria. High levels of recovery of spiked bacteria were seen, particularly at low concentrations. The examples of a calculated recovery of >100% are attributed to the variability in Ct values used to generate these values. An additional explanation is the use of estimated counts in killed bacterial suspensions to derive spiked numbers of bacteria.
Amplification was only observed in the plasma fraction of blood. Phagocytosis of bacteria by leukocytes and subsequent concentration in the buffy coat has been reported, 21 but improved sensitivity of PCR has also been observed in human serum compared to whole blood samples.20,32 The experimental goal of the current study was to show the possibility of amplification from blood and blood components, and killed bacteria were used in the experiments for reasons of availability and safety of laboratory personnel. The in vitro partitioning of killed bacteria in blood may differ from that of live bacteria; additional studies may be warranted.
When used with field-collected tissues, the assay described in the present study agrees with results of microbial culture, the “gold standard,” in the majority of cases. The calculated diagnostic sensitivity may not reflect true utility of this test to detect Brucella. In cases where PCR results were negative in the face of a positive culture, discrepancies may be explained by differences in sampling of potentially low-level or focal infections, as homogenates of large tissue blocks (approximately 5 g) were used for culture, while DNA was extracted from 10 to 25 mg of solid tissue. Field samples found to be PCR-positive but culture-negative were frequently from animals with other culture- or PCR-positive tissues, suggesting not “false positives,” but infection not detected by culture. Amounts of bacterial DNA detected in PCR-positive and culture-negative tissues were generally low, close to the LOD. Suboptimal or prolonged storage of frozen tissues may have decreased bacterial viability, limiting successful culture. Alternately, real false positives may have occurred from cross-contamination of tissues with bacteria during necropsy tissue collection or storage.
Blood cultures were not performed in large numbers of field cases tested in the current study. No blood or blood component was positive by PCR by the assay. Only 2 culture-positive animals had blood and tissues submitted, and both individuals were negative. Most of the blood samples came from wild, presumptively healthy animals, which would not be expected to have bacteremia. Fecal culture for Brucella was problematic due to overgrowth by fecal flora and was not pursued further. Four animals positive by PCR and culture of tissues were also positive by fecal PCR, but fecal shedding was not consistently seen. The assay also successfully amplified Brucella DNA from culture-positive, paraffin-embedded tissues, although detection was most successful when multiple paraffin blocks from the same animal were tested.
Comparison of the technique described herein with serology was not possible because very small numbers of submissions to the current study included both serum and tissues from an individual animal. While serology has clear utility in assessing rates of exposure to Brucella in populations, known deficits of serologic testing underline the necessity for direct identification of Brucella sp. by either culture or PCR for definitive diagnosis in an individual case.
In summary, the described method is a sensitive, specific, real-time PCR technique for detection of Brucella spp. bacteria in a variety of sample matrices. The assay has the benefit of improved speed and safety compared to culture and identifies the direct presence of the organism. Although intended and validated for use with marine mammal tissues and marine-origin Brucella spp., previous studies have shown utility of this primer-probe combination for all recognized species of Brucella. Field use of this assay indicates that replicate testing of multiple tissues from an individual provides the most accurate evaluation of infection status.
Footnotes
Acknowledgements
This is contribution #175 from the Sea Research Foundation. The authors would like to thank Darla Ewalt and Christine Quance from the National Veterinary Services Laboratory; Thomas Ficht of Texas A&M University for his donation of reagent; Guillermo Risatti and Andrew Draghi II, Department of Pathobiology and Veterinary Science, University of Connecticut, for development assistance; Jessica Hoag and Carolina Ruiz, interns at the Mystic Aquarium & Institute for Exploration; Mary Schwab of the Molecular Pathology Laboratory, Dartmouth Medical School and Dartmouth Hitchcock Medical Center; Amy Smith of Qiagen Inc. for technical support; and Edmund Kadyszewski for statistical advice. Lastly, the authors would like to acknowledge the many collaborators from private and public institutions and organizations who provided tissue samples for this study, including Alaska Department of Fish and Game; Alaska Sea Life Center; California Department of Fish and Game; Cape Cod Stranding Network; Colorado State University; Tribal Community of St. Paul Island, Alaska; Department of Wildlife Management, North Slope Borough, Barrow, Alaska; Georgia Aquarium; Harbor Branch Oceanographic Institute; Hubbs Sea World Research Institute; John G. Shedd Aquarium; Marine Animal Lifeline; Maryland Department of Natural Resources; Mirage Aquarium; Mote Marine Laboratory; Mystic Aquarium; National Aquarium; National Marine Mammal Laboratory; National Ocean Service; New England Aquarium; NOAA Center for Marine Animal Health; Riverhead Foundation; Roger Williams Park Zoo; Sea Life Park; SeaDoc Society; Seattle Aquarium; The Marine Mammal Center; University of New England; U.S. Navy Marine Mammal Program; Vancouver Aquarium; Virginia Aquarium Stranding Program; Washington Department of Fish and Wildlife; and Woods Hole Oceanographic Institution.
a.
Brucella pinnipedialis, accession no. 311585, National Veterinary Services Laboratory, Ames, IA.
b.
Brucella ceti str. Cudo (GenBank taxonomy ID 595497), T. Ficht, Texas A&M University, College Station, TX. 20
c.
Brucella abortus complement fixation test antigen, National Veterinary Services Laboratory, Ames, IA.
d.
Connecticut Veterinary Medical Diagnostic Laboratory, Storrs, CT.
e.
Ultrospec 1000 UV/Visible Spectrophotometer, Pharmacia BioTech, Piscataway, NJ.
f.
DNeasy Blood & Tissue Kit, Qiagen Inc., Valencia, CA.
g.
Kendall Monoject #8881311149 (tripotassium EDTA) and #8881320157 (sodium heparin), Covidien, Mansfield, MA.
h.
ZR Fecal DNA Kit, Zymo Research, Orange, CA.
i.
MWG Biotech, High Point, NC.
k.
HotStarTaq, Qiagen Inc., Valencia, CA.
l.
Zymoclean Gel DNA Recovery Kit, Zymo Research, Orange, CA.
m.
A-Addition and PCR CloningPlus Kits, Qiagen Inc., Valencia, CA.
n.
Plasmid Maxiprep Kit, Qiagen Inc., Valencia, CA.
q.
BLASTn, National Center for Biotechnology Information, U.S. National Library of Medicine, Bethesda, MD.
r.
7300 Real-time PCR System, Applied Biosystems, Foster City, CA.
s.
QuantiTect Multiplex NoROX Mastermix, Qiagen Inc., Valencia, CA.
t.
Statistix for Windows (version 8.0), Analytical Software, Tallahassee, FL.
u.
SAS version 8.2, SAS Institute Inc., Cary, NC.
v.
Computer Programs for Epidemiologists (PEPI) version 4.0, Sagebrush Press, Salt Lake City, UT.
w.
Invitrogen Corp., Carlsbad, CA.
x.
Stomacher 80 Lab Blender, Seward Laboratory Systems, London, England.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article:
This work was supported by the National Oceanic and Atmospheric Administration Oceans and Human Health Initiative grant (NA04OAR4600209) and by a Bernice Barbour Foundation grant (#000342006).