
Abstract
A total of 23 clinical isolates of Fusobacterium spp. were recovered at necropsy over a 2-year period from the respiratory tract of white-tailed deer (Odocoileus virginianus). Isolates were identified as Fusobacterium varium (18/23), Fusobacterium necrophorum subsp. funduliforme (3/23), and Fusobacterium necrophorum subsp. necrophorum (2/23). Using polymerase chain reaction–based detection of virulence genes, all F. necrophorum isolates were positive for the promoter region of the leukotoxin operon and the hemagglutinin-related protein gene, while all F. varium isolates were negative. The presence of the leukotoxin gene in F. necrophorum isolates and the absence of this gene in F. varium isolates were confirmed by Southern hybridization using 2 separate probes. Toxicity to bovine polymorphonuclear leukocytes was observed with all F. necrophorum isolates, but was not observed in any F. varium isolates. Susceptibility to antimicrobials was markedly different for F. varium as compared to F. necrophorum. In summary, no evidence of leukotoxin production was detected in any of the 23 F. varium isolates used in the current study. The data suggests that F. varium, the most common species isolated, may be a significant pathogen in deer with a different virulence mechanism than F. necrophorum.
Keywords
Introduction
Fusobacterium spp. are Gram-negative, non–spore-forming, anaerobic, rod-shaped bacteria of the Bacteroidaceae family. Fusobacterium necrophorum, which is currently recognized as 2 subspecies, F. n. subsp. necrophorum and F. n. subsp. funduliforme, is one of the most common anaerobic bacteria isolated from abscesses, respiratory tract infections, and other necrotizing infections in domestic livestock, wild mammals, and human beings.6,15,23 While the role of F. necrophorum in hepatic abscessation, necrotizing laryngitis, and foot rot in cattle, small ruminants, and camelids is well recognized, the association with severe respiratory tract infections in deer is infrequently recognized in the literature and the pathogenesis is poorly characterized.3,21 Although less extensively investigated, Fusobacterium varium has been associated with ulcerative colitis, colonic neoplasia, decubitus ulcers, and respiratory tract infections in human beings, and with suppurative or ulcerative lesions of the gastrointestinal tract or oral cavity in animals.4,11,19 Various Fusobacterium spp. have been isolated from wild ruminants affected by necrobacillosis, a common disease of farm-raised deer characterized by purulent necrotic lesions most commonly affecting the mouth, pharynx, lung, liver, or feet.7,21 Some of the most frequently isolated pathogens in necrobacillosis cases are F. necrophorum and F. varium, with coinfections commonly associated with Trueperella (Arcanobacterium) pyogenes.4,8 The infection often spreads to multiple organs resulting in death, as antibiotic therapy is often not successful. 12 The objective of the current study was to phenotypically and genotypically characterize clinical isolates of Fusobacterium spp. from the respiratory tract of white-tailed deer (Odocoileus virginianus).
Materials and methods
Collection and identification of isolates
Twenty-three clinical isolates of Fusobacterium spp. were used in the current study. All isolates were obtained at necropsy at the Animal Diagnostic Laboratory at The Pennsylvania State University (University Park, Pennsylvania) from the respiratory tract (lungs or larynx) of white-tailed deer from Pennsylvania. The selected isolates were presumptively identified as Fusobacterium sp. and further analyzed as described below (Table 1). Additionally, 3 reference strains, F. varium (American Type Culture Collection [ATCC] 8501), F. n. subsp. necrophorum (ATCC 25286), and F. n. subsp. funduliforme (ATCC 51357), were included in the study. a
Identification of Fusobacterium spp. isolates from white-tailed deer (Odocoileus virginianus).*

Kit 1 (Rapid ANAII, Remel Inc., Lenexa, KS) and kit 2 (RapID 32A, bioMérieux SA, Marcy l’Etoile, France). ID = identification; rDNA = ribosomal DNA; Sed = sedimentation in broth; Lip = lipase activity; Bile = bile tolerance. Numbers in parentheses are quality of ID percentage.
Highest quality ID (72%) for Clostridium haemolyticum was inconsistent with negative Gram staining. Result shown is second highest quality ID.
Tissue samples were cultured on prereduced Brucella agar plates b supplemented with 5% sheep blood, hemin (5 mg/l), and vitamin K (10 mg/l), and then incubated at 37°C for 48 hr in an anaerobic chamber. The 3-way antibiotic disk diffusion susceptibility test and nitrate test were conducted on Brucella agar plates with vancomycin, kanamycin, colistin, and nitrate disks. c Nitrate-negative isolates showing resistance to vancomycin and susceptibility to kanamycin and colistin were presumptively determined to be Fusobacterium sp. 9 Following presumptive identification, the indole test was performed by placing 1 drop of indole reagent c onto an antibiotic disk. A pre-reduced McClung–Toabe egg yolk agar plate b was inoculated and incubated anaerobically at 37°C for 48–72 hr to determine lipase activity. Bile tolerance was determined by plating colonies onto pre-reduced Bacteroides fragilis isolation agar plates b containing 20% bile and incubating anaerobically at 37°C for 48–72 hr. Overnight cultures in prereduced anaerobically sterilized brain–heart infusion broth were observed for growth and sedimentation. All isolates were analyzed with commercial kitsd,e according to the manufacturers’ instructions.
DNA extraction and 16S ribosomal DNA sequencing
Chromosomal DNA was extracted from all isolates. Briefly, cultures were grown anaerobically on pre-reduced blood agar plates at 37°C for 48–72 hr. The DNA was isolated using a commercial kit f according to the manufacturer’s instructions. The extracts were stored at −20°C. Universal primers (p515FPL forward GCGGATCCTCTAGACTGCAGTGCCAGCAGCCGCGGTAA, and p13B reverse CGGGATCCCAGGCCCGGGAACGTATTCAC) were used to amplify a region of the 16S ribosomal RNA gene by polymerase chain reaction (PCR). 22 Amplified products were electrophoresed in 1.5% agarose gels and purified using a commercial kit. g The purified PCR product was submitted to a commercial laboratory h for sequence analysis. For each isolate, sequences with >99% agreement were calculated using commercially available software i and selected for further analysis. Sequence alignment and phylogenetic analysis were conducted using Web-based software j and the neighbor-joining method. 24 Partial 16S ribosomal DNA (rDNA) sequences of all clinical isolates from the present study were deposited in GenBank and assigned accession numbers KC407916 through KC407943.
Lkt promoter and hemagglutinin gene PCR
Primers specific for the promoter region of the lktB gene and the hemagglutinin-related protein gene were used. The “lktpXmXh” primers detect the lkt promoter in both subspecies, whereas the “fund” primers detect the lkt promoter of only the funduliforme subspecies. 27 The “haem” primer detects the gene that codes for hemagglutinin-related protein. 1 Primers were as follows: lktpXmXh forward 5′-TCTCCCGGGCTCGAGGAAATCTTTAAAGCAC-3′, lktpXmXh reverse 5′-TCTCCCGGGCATAATTTCTCCCAATTTTATT-3′, fund forward 5′-CTCAATTTTTGTTGGAAGCGAG-3′, fund reverse 5′-CATTATCAAAATAACATATTTCTCAC-3′, haem forward 5′-CATTGGGTTGGATAACGACTCCTAC-3′, and haem reverse 5′-CAATTCTTTGTCTAAGATGGAAGCGG-3′.1,27 Amplification was performed in a thermal cycler using a modification of previously described conditions. 27 The PCR conditions were as follows: initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 45°C for 30 sec, and 62.4°C for 30 sec, extension at 72°C for 1 min, and the final extension at 72°C for 4 min. The annealing temperature was reduced from 62.4°C for lktpXmXh primers to 55.1°C for haem primers and 50.8°C for fund primers. The amplified products were electrophoresed in a 1.5% agarose gel containing ethidium bromide.
Leukotoxin gene detection by Southern hybridization
Southern blot hybridization was performed as previously described with modifications. 17 Briefly, chromosomal DNA was isolated using a commercial kit k as per the manufacturer’s instructions. Chromosomal DNA (1.5 μg) was digested to completion using HaeIII and electrophoresed in a 1% agarose gel overnight. The resolved DNA fragments were transferred to a positively charged nylon membrane and immobilized by baking. Two DNA probes were prepared using a PCR-based digoxigenin (DIG)-labeled probe synthesis kit l following the manufacturer’s instructions. One probe was 1,200 base pairs (bp) and corresponded to a conserved lktA gene region of F. necrophorum. The second probe was 571 bp and corresponded to the promoter region of the lktA gene of F. necrophorum. The immobilized DNA was probed at 43.6°C for the 1,200-bp probe, and 40°C for the 571-bp probe. Colorimetric detection of hybridized signals was accomplished following the manufacturer’s instructions.
Leukotoxicity assay by flow cytometry
Leukotoxicity was determined by incubation of bovine polymorphonuclear leukocytes (PMNs) with sterile culture supernatant and propidium iodide (PI) as previously described. 16 Fusobacterium isolates were grown in prereduced anaerobically sterilized brain–heart infusion at 37°C to an absorbance of 0.7–0.8 at 600 nm, placed on ice, and pelleted by centrifugation at 10,000 × g for 10 min at 4°C as previously described. 25 Supernatants were filter sterilized with 0.22-µm membrane filters and stored at −80°C. Bovine peripheral PMNs were prepared as previously described. 25 Purified leukocytes were washed twice and resuspended in 0.01 M phosphate buffered saline. Viable cell concentration was determined with a hemocytometer by the trypan blue dye exclusion method. 25 Approximately 2 × 106 PMNs were placed into sterile 5-ml polystyrene culture tubes and incubated with an equal volume of filter-sterilized culture supernatant. Cells were incubated for 45 min at 37°C and 5% CO2, washed twice, and resuspended in phosphate buffered saline. Five microliters of 1 mg/ml PI was added to each tube and the tubes incubated at room temperature for 5–10 min in the dark. Samples were analyzed on a flow cytometer. m The proportion of cells stained with PI was determined by the detection of red fluorescence of cells in the PMN gate.
Antimicrobial susceptibility determination
The broth microdilution method of antimicrobial susceptibility was performed by a commercial system n according to the manufacturer’s instructions. Colonies were selected from 48-hr cultures on blood agar plates, inoculated into Mueller–Hinton broth tubes, n and transferred into supplemented Brucella broth tubes n adjusted to 1 × 106 colony forming units/ml. Aliquots (100 µl) were transferred to wells of a 96-well antimicrobial susceptibility plate. o Plates were covered by perforated adhesive seals, incubated anaerobically at 37°C for 48 hr, and the minimal concentration of antimicrobial agents that inhibited bacterial growth was determined by visual examination. Breakpoints were interpreted by Clinical and Laboratory Standards Institute (CLSI) guidelines. 26 Because no CLSI guidelines are available for Fusobacterium spp., the CLSI breakpoints for other veterinary pathogens were used (Table 2). Specifically, minimal inhibitory concentration (MIC) breakpoints for bovine respiratory disease pathogens were used when available, otherwise MIC breakpoints for swine respiratory pathogens, Enterobacteriaceae, or undefined veterinary pathogens were used. 26 No CLSI-approved MIC breakpoints were available for neomycin or tylosin, therefore breakpoints were obtained from the product data sheet provided by the manufacturer n for Gram-negative bacteria.
Antimicrobial susceptibility of Fusobacterium spp. from white-tailed deer (Odocoileus virginianus).*
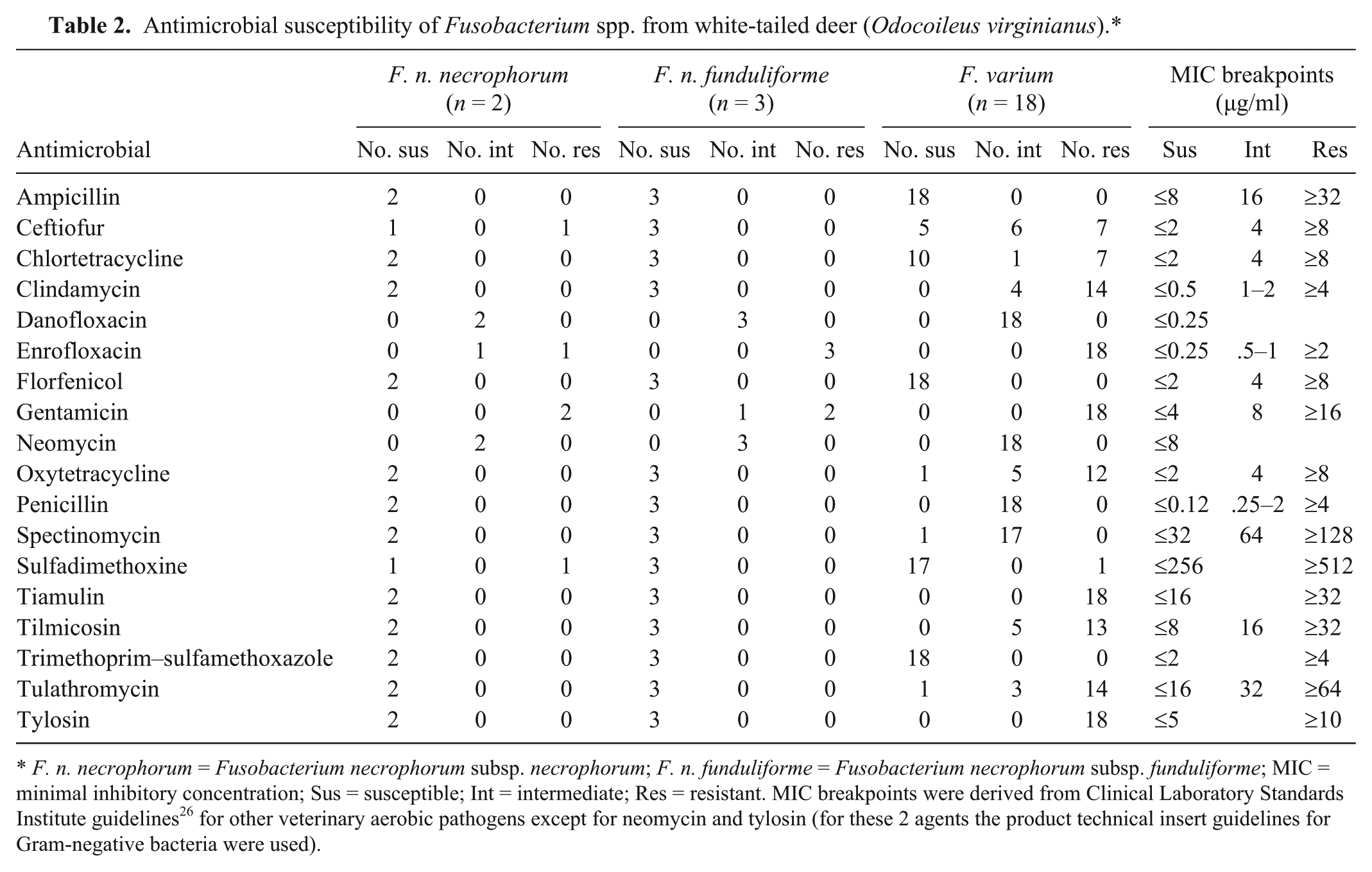
F. n. necrophorum = Fusobacterium necrophorum subsp. necrophorum; F. n. funduliforme = Fusobacterium necrophorum subsp. funduliforme; MIC = minimal inhibitory concentration; Sus = susceptible; Int = intermediate; Res = resistant. MIC breakpoints were derived from Clinical Laboratory Standards Institute guidelines 26 for other veterinary aerobic pathogens except for neomycin and tylosin (for these 2 agents the product technical insert guidelines for Gram-negative bacteria were used).
Statistical analysis
Flow cytometry data was evaluated by one-way analysis of variance with comparisons for each pair using the Student t-test. Standard statistical software p was used. For all analyses, a value of P < 0.05 was considered significant.
Results
Species identification of isolates
A single Fusobacterium sp. was isolated from each animal included in the study. The isolates included 18 (78.3%) F. varium, 3 (13.0%) F. n. subsp. funduliforme, and 2 (8.7%) F. n. subsp. necrophorum. Results are summarized in Table 1. Determination of species or subspecies was made by standard phenotypic methods and analysis of biochemical characteristics through the use of 2 commercial kits with confirmation by 16S rDNA sequence analysis. The kits performed similarly in the identification of F. n. subsp. necrophorum; however, F. n. subsp. funduliforme was incorrectly identified as F. nucleatum by kit 2 e and some isolates of F. varium were incorrectly identified as F. nucleatum by kit 1. d The sequences of the 5 F. necrophorum isolates clustered together and the 2 subspecies were not distinguishable by 16S rDNA sequences alone (Fig. 1). All F. necrophorum isolates also clustered apart from the previously reported F. equinum sequences, thus excluding the potential misidentification of F. necrophorum as F. equinum. 28 Similarly, all F. varium isolates clustered apart from F. necrophorum. Previously deposited sequences for F. nucleatum and F. equinum were distinct from the clinical isolates evaluated in the current study.
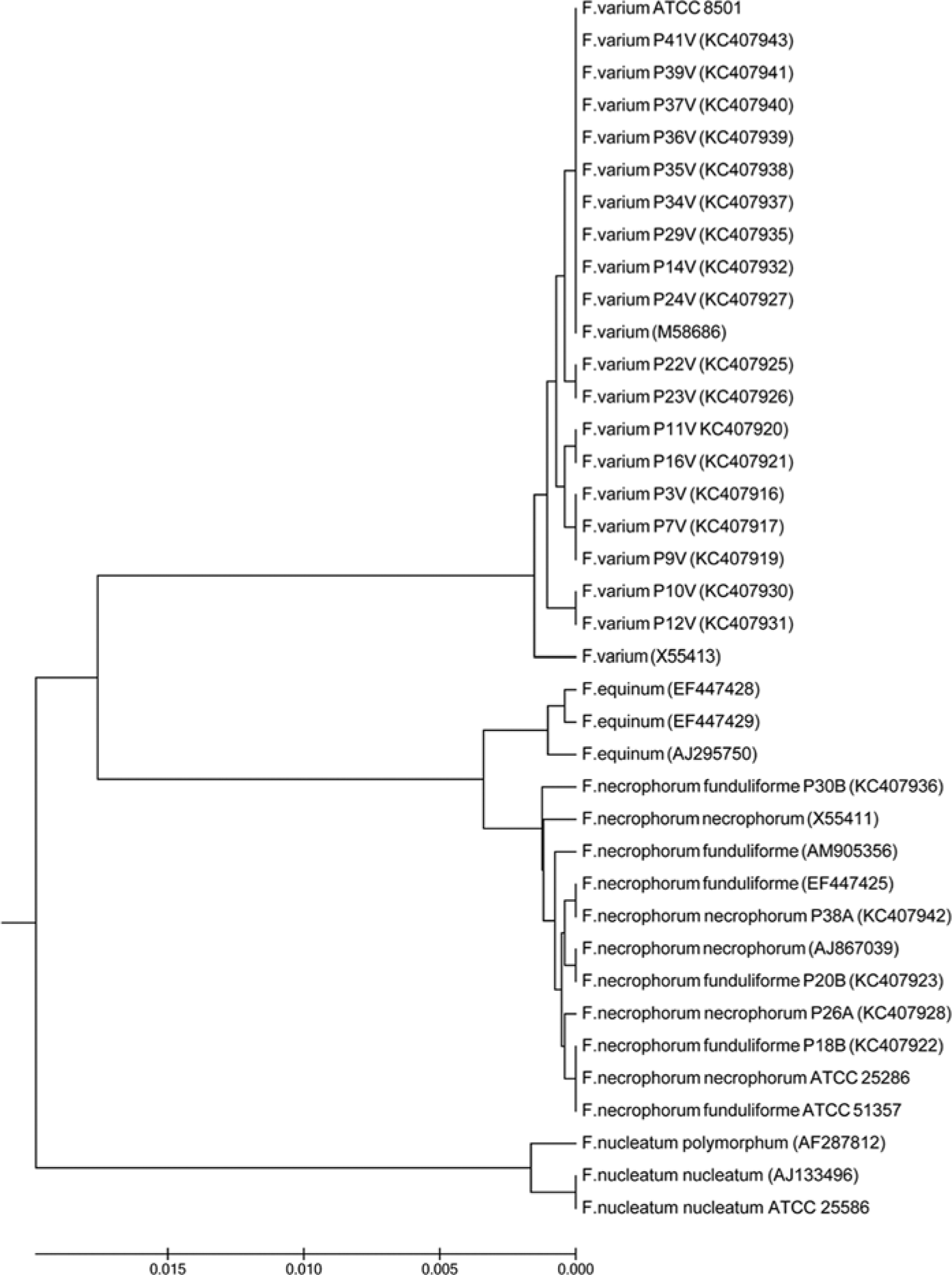
Phylogenetic analysis (neighbor-joining method) of the 16S ribosomal RNA gene sequences of clinical Fusobacterium spp. isolates aligned with previously deposited Fusobacterium spp. sequences.
Virulence gene analysis
Using the lktp primers for the promoter region of lktB, all F. varium isolates were negative for the lkt promoter. Using these same primers, all F. n. subsp. necrophorum isolates amplified the expected product of approximately 571 bp, while all F. n. subsp. funduliforme isolates amplified a slightly smaller product of approximately 449 bp as expected. With the fund primers specific for the lkt promoter region of F. n. subsp. funduliforme, all F. varium isolates were negative. With these same primers, all F. n. subsp. necrophorum were negative and 1 out of 3 (33%) F. n. subsp. funduliforme isolates amplified the expected 337-bp product. Using the haem primers for the hemagglutinin-related protein gene, all F. varium isolates were negative. With the haem primers, all F. n. subsp. necrophorum strains amplified the expected 311-bp product, while all F. n. subsp. funduliforme isolates were negative.
Using 2 different probes for the detection of the lktA gene and the leukotoxin promoter, it was determined by Southern hybridization that the leukotoxin gene was absent from all isolates of F. varium and present in all isolates of F. necrophorum. When probed with the 1,200-bp DIG-labeled F. n. subsp. necrophorum lktA, isolates of F. n. subsp. necrophorum produced a hybridizing band (approximately 23 kb), and F. n. subsp. funduliforme produced a hybridizing band of a slightly larger size (Fig. 2A). When probed with the 571-bp DIG-labeled F. n. subsp. necrophorum lkt promoter, isolates of F. n. subsp. necrophorum produced a hybridizing band (approximately 23 kb), and F. n. subsp. funduliforme isolates showed no hybridization (Fig. 2B). No F. varium isolates produced a hybridizing band with either probe, indicating the absence of the leukotoxin gene.
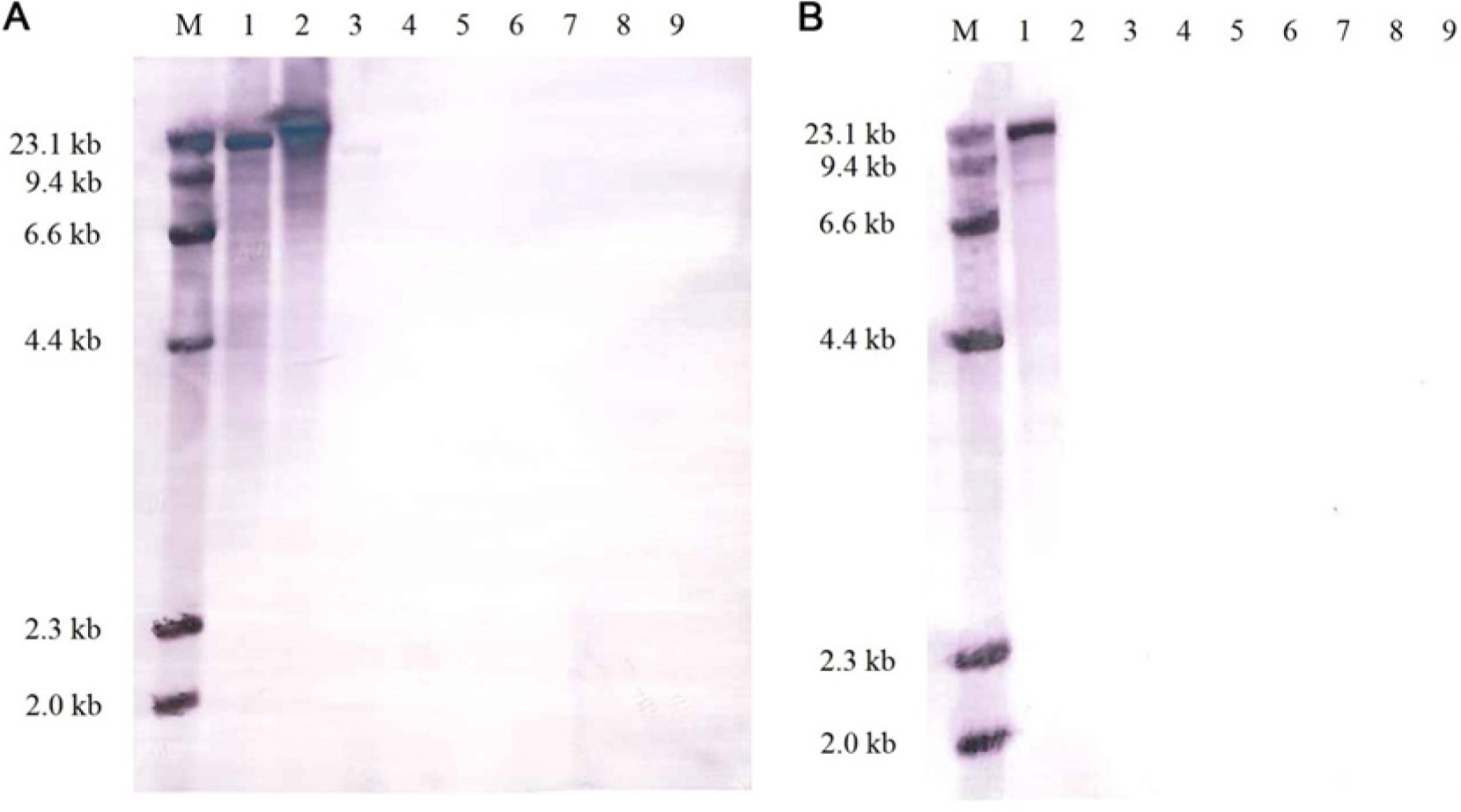
Southern hybridization patterns of HaeIII-digested genomic DNA probed with digoxigenin (DIG)-labeled Fusobacterium necrophorum lktA gene conserved region (
Leukotoxin activity by flow cytometry
The toxicity of culture supernatant to bovine PMNs, as determined by PI staining, was highly dependent on fusobacterial species (Fig. 3). The uptake of PI by PMNs was highest in cells incubated with supernatant of F. n. subsp. necrophorum (mean: 67.0%; standard error [SE]: 11.5), intermediate with F. n. subsp. funduliforme (mean: 18.9%; SE: 9.1), and very low for F. varium (mean: 2.3%; SE: 0.1), which approximated the negative control (mean: 1.2%; SE: 0.4). Leukotoxicity, as determined by mean PI uptake, for F. n. subsp. necrophorum differed significantly from the means of F. n. subsp. funduliforme (P < 0.0001) and F. varium (P < 0.0001). Leukotoxicity for F. n. subsp. funduliforme differed significantly from F. n. subsp. necrophorum (P < 0.0001) and from the mean of F. varium (P < 0.002). The means of F. varium and the negative control were not significantly different (P > 0.05).
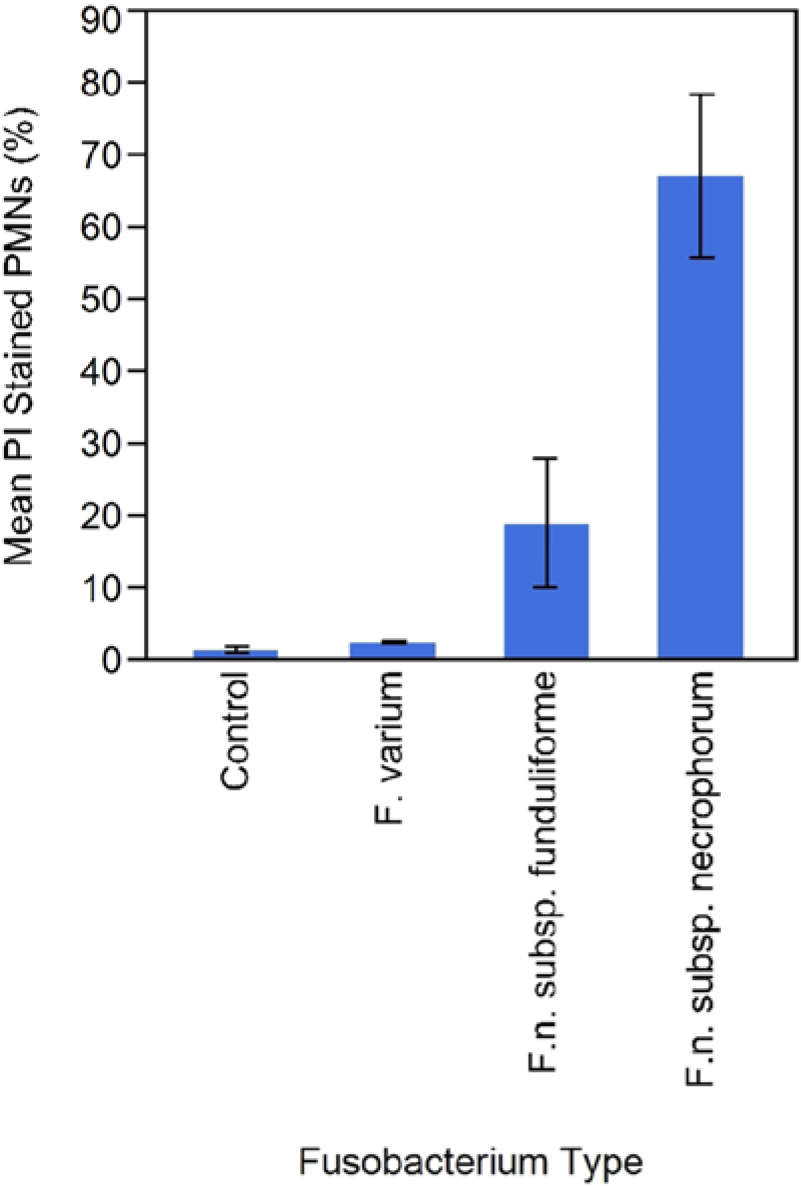
Leukocyte viability assay. Bovine polymorphonuclear leukocytes (PMNs) treated with culture supernatant were stained with propidium iodide (PI) and analyzed by flow cytometry. Bars represent means for each species; brackets represent standard error. Control = no culture supernatant.
Antimicrobial susceptibility
The results of the broth microdilution antimicrobial susceptibility tests are summarized in Table 2. While F. n. subsp. necrophorum and F. n. subsp. funduliforme showed similar patterns of susceptibility, the susceptibility of F. varium was markedly different for many antimicrobials. All isolates (23/23) of all 3 fusobacterial species were susceptible to ampicillin, florfenicol, and trimethoprim–sulfamethoxazole. Fewer F. varium isolates were susceptible to chlortetracycline, clindamycin, oxytetracycline, tiamulin, tilmicosin, tulathromycin, and tylosin as compared with F. necrophorum. Antimicrobials to which isolates demonstrated intermediate or variable susceptibilities included ceftiofur, danofloxacin, neomycin, penicillin, spectinomycin, and sulfadimethoxine. Nearly all isolates (22/23) were resistant to enrofloxacin and gentamycin.
Discussion
Respiratory tract infection is one of the primary causes of mortality in farm-raised white-tailed deer.3,8 The definitive cause of such infections remains undetermined, but one of the most commonly isolated pathogens is Fusobacterium.4,8 It is important for the deer farming industry and the animal health community that the etiology and pathogenesis of this disease are more fully characterized in order to develop appropriate treatment and prevention measures. The current study shows that F. varium comprises a large proportion of the clinical isolates from cases of fusobacterial respiratory tract infections in deer. Standard phenotypic methods with the inclusion of lipase activity, bile tolerance, and sedimentation were sufficient for subspecies level identification of the clinical Fusobacterium isolates used in the current study. However, such methods are labor intensive and time consuming and may be insufficient for speciating or subtyping some Fusobacterium spp. isolates. Commercial bacterial identification kits were useful as part of a diagnostic scheme, but were inadequate as the sole means of identification of many Fusobacterium spp. to the species or subspecies level. Although F. n. subsp. necrophorum was consistently correctly identified at the species level with both test kits, F. n. subsp. funduliforme and F. varium were sometimes misidentified as F. nucleatum. Genotypic analysis by PCR or ribosomal gene sequencing may be preferable for diagnosis or confirmation.
Fusobacterium necrophorum possesses several virulence factors, and leukotoxin is considered to be the major virulence factor involved in animal infections.15,23 Virulence factors of F. varium, however, have not been well characterized. The F. necrophorum leukotoxin operon consists of 3 genes, lktB, lktA, and lktC corresponding respectively to an outer membrane protein with putative transporter function, the leukotoxin protein, and a protein of unknown function. 23 The lkt operon promoter region is distinct for each of the 2 subspecies of F. necrophorum and allows for differentiation of the 2 subspecies. 27 Amplification by PCR of the lkt promoter and the hemagglutinin gene differentiated F. necrophorum subspecies from other Fusobacterium spp. 10 In the current study, the inconsistency in the band produced by F. n. subsp. funduliforme with fund primers may represent polymorphisms in the sequence of this virulence gene.
No evidence of the leukotoxin gene or leukotoxin production by any isolate of F. varium was detected by PCR analysis, Southern blotting, or the leukocyte viability assay. Although it is fundamentally difficult to prove the absence of a gene, 2 the sum of these analyses collectively supports the conclusion that F. varium does not produce leukotoxin. It is, therefore, possible that the pathogenicity of F. varium is not associated with leukotoxin production but with some other virulence factor. One study suggests that the leukotoxin gene is much less prevalent in non-bovine, invasive animal and human strains and, thus, should not be considered the primary virulence factor in all hosts. 13 For example, lysis of erythrocytes by hemolysin has been proposed as a major fusobacterial virulence factor. 14 Additionally, butyric acid has been identified as a potential virulence factor in F. varium based on cytotoxicity to Vero (African green monkey kidney epithelial) cells and production of colonic lesions in mice.18,19 Butyric acid has been demonstrated to induce apoptosis of colonic epithelium and to induce secretion of inflammatory cytokines. 19 Under certain conditions, some commensal organisms such as F. varium may be capable of invading epithelial cells and activating early host inflammatory responses. 19
Few studies on antimicrobial susceptibility of F. varium have been reported, although the susceptibility of other Fusobacterium species has been investigated. The results of the current study suggest that susceptibility to antimicrobials is markedly different among the Fusobacterium species evaluated, and are consistent with previous studies.5,11,20 It has been reported that most Fusobacterium spp. are sensitive to β-lactams and clindamycin, but that F. varium tends to be more resistant. 11 The data of the current study supports the increased resistance of F. varium to clindamycin as well as chlortetracycline, oxytetracycline, tiamulin, tilmicosin, tulathromycin, and tylosin in comparison to F. necrophorum. This finding is of clinical relevance to managers of captive deer herds and wild ruminant populations because traditional treatment and prevention efforts have been directed toward F. necrophorum infections, selecting antibiotics and formulating vaccines accordingly, as most fusobacterial infections in animals have been attributed to F. necrophorum. However, in the dataset used in the current study, the majority (78.3%) of clinical isolates were F. varium, which suggests that most cases of necrobacillosis may be caused by a different species of Fusobacterium with a different antibiotic resistance profile and, potentially, a different antigenic profile. Therefore, in order to properly diagnose and provide critical information for treatment and preventive measures, it is essential that clinical Fusobacterium isolates are identified to the species or subspecies level. Additional studies are required to determine virulence factors of F. varium and the pathogenicity and host immune response to F. varium infection in order to develop effective treatment and prevention strategies.
Footnotes
Acknowledgements
The authors thank the Missouri Whitetail Breeders and Hunting Ranch Association and Drs. Vivek Kapur, Arthur Hattel, Jenny Fisher, and Subhashinie Kariyawasam, as well as Valerie Linter of The Pennsylvania State University, Department of Veterinary and Biomedical Sciences for their assistance.
a.
American Type Culture Collection, Manassas, VA.
b.
Remel Inc., Lenexa, KS.
c.
Anaerobe Systems, Morgan Hill, CA.
d.
Rapid ANAII, Remel Inc., Lenexa, KS.
e.
RapID 32A, BioMérieux SA, Marcy l’Etoile, France.
f.
DNeasy Blood and Tissue Kit, Qiagen GmbH, Hilden, Germany.
g.
QIAquick Gel Extraction Kit, Qiagen GmbH, Hilden, Germany.
h.
Davis Sequencing, Davis, CA.
i.
Vector NTI Advance 11.0 Align X, Invitrogen Corp., Carlsbad, CA.
k.
Wizard genomic DNA purification kit, Promega Corp., Madison, WI.
l.
Roche Diagnostics Corp., Indianapolis, IN.
m.
Cytomics FC500, Beckman Coulter, Fullerton, CA.
n.
Sensititre System, ThermoFisher Scientific; formerly TREK Diagnostics; Cleveland, OH.
o.
BOPO6F, ThermoFisher Scientific; formerly TREK Diagnostics; Cleveland, OH.
p.
JMP, SAS Institute Inc., Cary, NC.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.