
Abstract
Staphylococcus pseudintermedius is a commensal and opportunistic pathogen of dogs. It is mainly implicated in canine pyoderma, as well as other suppurative conditions of dogs. Although bacterial culture is routinely used for clinical diagnosis, molecular methods are required to accurately identify and differentiate S. pseudintermedius from other members of the Staphylococcus intermedius group. These methods, owing largely to their cost, are not easy to implement in nonspecialized laboratories or veterinary practices. In the current study, loop-mediated isothermal amplification (LAMP), a novel isothermal nucleic acid amplification procedure, was employed to develop a rapid, specific, and sensitive S. pseudintermedius assay. Different detection strategies, including the use of a lateral flow device, were evaluated. The assay was evaluated for cross-reactivity against 30 different bacterial species and validated on a panel of 108 S. pseudintermedius isolates, originating from different dog breeds and locations within the United Kingdom. The assay was specific, showing no cross-reactivity during in silico and in vitro testing. When tested using DNA extracts prepared directly from 35 clinical surgical site swabs, the assay could detect S. pseudintermedius in less than 15 min, with a diagnostic sensitivity of 94.6%, superior to that of a polymerase chain reaction method. The LAMP assay also had an analytical sensitivity in the order of 101 gene copies, and the amplified products were readily detected using a lateral flow device. The LAMP assay described in the present study is simple and rapid, opening up the possibility of its use as a diagnostic tool within veterinary practices.
Introduction
Staphylococcus pseudintermedius is a Gram- and coagulase-positive bacterium, which belongs to the Staphylococcus intermedius group (SIG), a group which consists of S. intermedius, S. pseudintermedius, and S. delphini. Staphylococcus pseudintermedius naturally colonizes the skin, coat, nose, mouth, and anus of dogs, but can also become an opportunistic pathogen where it is implicated in wound infections, pyoderma, otitis externa, endometritis, and other suppurative conditions. 17 Methicillin-resistant strains of S. pseudintermedius are thought to be an important and emerging problem that poses a significant challenge to small animal veterinary medicine.8,17
Currently, routine diagnosis of S. pseudintermedius is performed using phenotypic methods. However, these methods, as well as being unable to accurately differentiate S. pseudintermedius from some other members of the SIG, can also be time consuming and expensive. This has meant that the use of molecular methods is essential.3,20 Automated systems, such as the use of matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry, have also been described for S. pseudintermedius identification. 6 Nonetheless, a review on S. pseudintermedius noted that despite rapid identification of S. pseudintermedius (and other bacteria) through the use of automated systems such as MALDI-TOF, the financial burden associated with these systems limits their use to large commercial diagnostic laboratories. 3
Loop-mediated isothermal amplification (LAMP) is a novel isothermal nucleic acid amplification method with high specificity, efficiency, and speed. It has been employed for the detection and identification of bacteria, viruses, and protozoa.7,12,14 The LAMP assay utilizes simple and inexpensive laboratory equipment and, as a result, has been explored as a potential molecular point-of-care diagnostic tool.5,16 Detection methods including the use of turbidity (based on magnesium pyrophosphate generation during the reaction), 12 fluorescent dyes (calcein and SYBR-1),4,19 and complexometric detectors, such as hydroxynaphthol blue, 9 have all been explored for the detection of LAMP reaction products. However, these detection methods have varying levels of complexity and user interpretation.
In the current study, a LAMP assay capable of rapid detection of S. pseudintermedius from DNA extracts prepared directly from clinical swabs was designed and evaluated. The method requires minimal instrumentation, which, when combined with a lateral flow device for detection, has the potential to give specific and unambiguous results in less than 30 min. Importantly, the assay could be used in veterinary practices as it has no requirements for a high level of technical expertise or expensive equipment.
Materials and methods
Samples for diagnostic specificity and analytical sensitivity testing
For evaluating the specificity, and diagnostic and analytical sensitivity of the assay, DNA extracts from 4 sets of samples (A–D) were used. Sets A, B, and C are summarized in Table 1. Set A consisted of 68 S. pseudintermedius isolates from routine sampling of dogs in the United Kingdom. These isolates were obtained from the University of Liverpool (UoL; Liverpool, United Kingdom), as pure cultures which were subcultured onto blood agar prior to DNA extraction. Sets B and C both came from Fitzpatrick Referrals (FPR; Godalming, Surrey, United Kingdom). Set B consisted of 40 clinical S. pseudintermedius isolates, previously cultured from surgical site infections (SSIs) in dogs admitted to FPR. These pure bacterial cultures were stored at −20°C and were subcultured on to blood agar prior to DNA extraction. Set C consisted of 35 clinical swabs from SSIs in dogs (FPR). Following inoculation on blood agar, mannitol salt agar, and MacConkey agar for routine biochemical analysis and identification, DNA was extracted directly from the swabs. All S. pseudintermedius DNA extracts used (sets A–C) were confirmed to be S. pseudintermedius by molecular analysis. Set D consisted of 30 non–S. pseudintermedius isolates (Table 2), obtained from the strain collection of the Animal Health and Veterinary Laboratories Agency (Weybridge, Surrey, United Kingdom) and were used to check the diagnostic specificity of the LAMP assay.
Summary of all the samples used for evaluating the specificity and diagnostic and analytical sensitivity of the loop-mediated isothermal amplification (LAMP) assay. The results of the use of the LAMP assay and the alternative molecular technique—the polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method—are also presented.
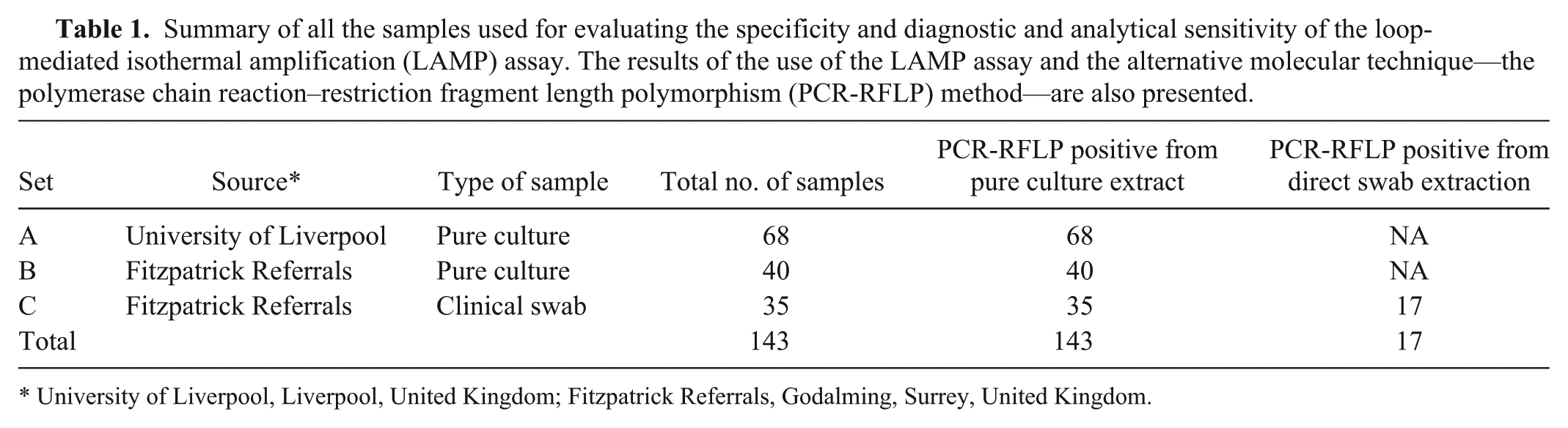
University of Liverpool, Liverpool, United Kingdom; Fitzpatrick Referrals, Godalming, Surrey, United Kingdom.
Bacteria isolates used in evaluating the specificity of the spsL loop-mediated isothermal amplification assay.

Field = fully characterized field isolates; ATCC = American Type Culture Collection.
DNA preparation
For DNA extraction from sets A, B, and D, approximately 5 colonies from stationary growth culture on blood agar were suspended in 1 ml of 0.1 M phosphate buffered saline (PBS), vortexed briefly, and then centrifuged at 13,400 × g for 5 min. The supernatant was discarded, and the bacteria were resuspended in 500 µl of nuclease-free water, before being heated to 95°C for 15 min. The cell debris was pelleted by centrifugation at 13,400 × g for 10 min. The crude lysates were used as template for LAMP and polymerase chain reaction (PCR).
DNA was extracted from swabs in set C by agitating them in 200 µl of 0.1 M PBS. Bacteria and transport media debris were pelleted by centrifugation for 2 min at 13,400 × g. The supernatant was discarded, and the pellet resuspended in 100 µl of nuclease-free water. Bacterial cells were lysed by heating to 95°C for 15 min. Cellular and transport media debris were subsequently pelleted by centrifugation for 2 min at 13,400 × g. The supernatant was stored at −20°C.
Molecular identification of S. pseudintermedius and DNA oligonucleotides
All S. pseudintermedius isolates used in the current study were identified using a PCR–restriction fragment length polymorphism (PCR-RFLP) method based on MboI restriction of the pta gene fragment, and was carried out as previously described. 2 A set of 6 primers (Table 3)—outer primers (F3 and B3), forward and backward inner primers (FIP and BIP) external, and loop primers (loopF and loopB) recognizing 8 distinct regions—were designed. a The primers were designed to target and amplify a region within the spsL gene of S. pseudintermedius ED99 (GenBank accession no. NC_017568). The spsL gene encodes one of the recently identified S. pseudintermedius cell wall anchor proteins, designated SpsL. 1
Characteristics of oligonucleotides used in the current study.*

The oligonucleotides were designed based on Staphylococcus pseudintermedius ED99 (NC_017568). F3 = forward external primer; B3 = back external primer; loopF = forward loop primer; loopB = back/reverse loop primer; FIP = forward internal primer; BIP = back internal primer.
F1c and F2, and B1c and B2 are co-synthesized to form FIP and BIP, which are used in the reaction, as opposed to the individual primers.
LAMP reaction
The 25-µl reaction mixture contained 15 µl of a commercial master mix, b 0.1 µM of each of the external primers, 0.2 µM of each of the loop primers, 2 µM of each of the internal primers (Table 3), 1 µl of template, and 6 µl of nuclease-free water. For real-time analysis, the LAMP reaction was performed on a real-time PCR thermocycler, with the following cycling conditions: 60 cycles of 30 sec at 65°C, followed by a 60-sec segment with stepwise increase of temperature from 65°C to 96°C. Fluorescence was measured in both segments, with the melting temperature of the amplicon calculated from the dissociation curve. A reaction was considered positive when the level of fluorescence crossed the critical threshold (CT), as calculated by the analysis software. c
For lateral flow device (LFD) detection of the amplified products, loopF and loopB primers were labeled at their 5′-ends with fluorescein isothiocyanate (FITC) and biotin, respectively. The reactions were incubated for 30 min (60 cycles) at 65°C. A commercial sandwich immunochromatographic LFD, d which captures only double-stranded amplicons containing both FITC and biotin, was used to detect the amplified product. Briefly, 1 µl of the LAMP product was diluted in 500 µl of running buffer, d mixed by pipetting, and 65 µl was added to the LFD. The result was read within 10 min.
For visualization of the reaction product on an agarose gel, 8-µl aliquots of the LAMP product were electrophoresed at 100 volts in a 2% Tris–acetate–ethylenediamine tetra-acetic acid (TAE) agarose gel and buffer. The gel was subsequently stained with ethidium bromide, and visualized under ultraviolet light.
Analytical specificity of the LAMP assay
The LAMP primers and expected amplicon (Fig. 1) were checked for cross-reactivity against gene sequences present on the National Center for Biotechnology Information (NCBI) database, using an online alignment search tool. e The cross-reactivity of the assay was further explored by testing the LAMP assay with DNA extracted from a panel of 30 different bacterial species (Table 2).

Amplicon of the spsL loop-mediated isothermal amplification assay, showing the designed primers, and their respective binding sites. The primers constitute a significant proportion of the amplicon.
Analytical sensitivity of the LAMP assay
A 325-bp fragment encompassing the target region of the LAMP assay was amplified by PCR using the F3 and B3 primers from the LAMP assay. For the PCR reaction, a 50-µl reaction contained 2 µl of DNA extract (from bacterial culture), 0.2 µM each of the F3 and B3 primers, and 25 µl of PCR master mix solution. f The reaction conditions were as follows: initial denaturation for 2 min at 94°C, followed by 30 cycles of 30 sec at 94°C, 30 sec at 55°C, 1 min at 72°C, and a final extension of 7 min at 72°C. The size of the PCR product was confirmed by electrophoresis of a 10-µl aliquot on 1% TAE agarose gel (90 V for 45 min). The PCR product was subsequently purified using a commercial PCR purification kit g as per the manufacturer’s instructions. The purified product was quantified by spectrophotometry, h with the number of gene copies present in 1 µl of the purified PCR product calculated from the spectrophotometer reading, using an online bioinformatics tool. i Prior to use, the template was serially diluted 1:5 in 10 mM Tris–HCl buffer and heated for 2 min at 95°C. Each dilution was tested in triplicate, with the limit of detection taken to be the concentration at which the target product was amplified in all 3 replicates.
Results
Microbiology results
Following culture and biochemical analysis, S. pseudintermedius was cultured from all 35 swabs in set C. In 16 of the 35 swabs, S. pseudintermedius only was cultured from the swabs, while the remaining 19 swabs yielded mixed cultures that included S. pseudintermedius.
PCR-RFLP analysis
PCR-RFLP analysis of all the isolates in sets A and B (108 S. pseudintermedius DNA extracts) resulted in the expected digestion profile. However, the expected digestion profile was observed only in 17 of the 35 DNA extracts from set C swabs, with no amplification or subsequent restriction enzyme digestion observed in any of the remaining 18 samples (Table 1).
Amplification of the spsL gene using LAMP
The expected sigmoidal logistic curve was only observed in reactions containing the target S. pseudintermedius DNA. In these reactions, amplification was confirmed after 7 min (14 cycles), with the reaction reaching its endpoint approximately 6 min later. No amplification was observed in the no-template control reactions, even after 60 cycles. Positive reactions with the LAMP assay progressed from the incubation start time to the reaction end point in less than 14 min. Spectrophotometric quantitation of the purified reaction product revealed that approximately 394 ng per µl of DNA was generated in 14 min of the reaction, from a starting template of approximately 100 gene copies. Furthermore, the propagation of a single product in the LAMP reaction was confirmed by melting curve analysis of the reaction. A single sharp dissociation peak at 86.7°C was observed only in the LAMP reactions containing S. pseudintermedius DNA. Furthermore, agarose gel electrophoresis of an aliquot of the reaction product revealed the characteristic ladder of multiple bands, only in the reactions containing S. pseudintermedius DNA extract (Fig. 2).
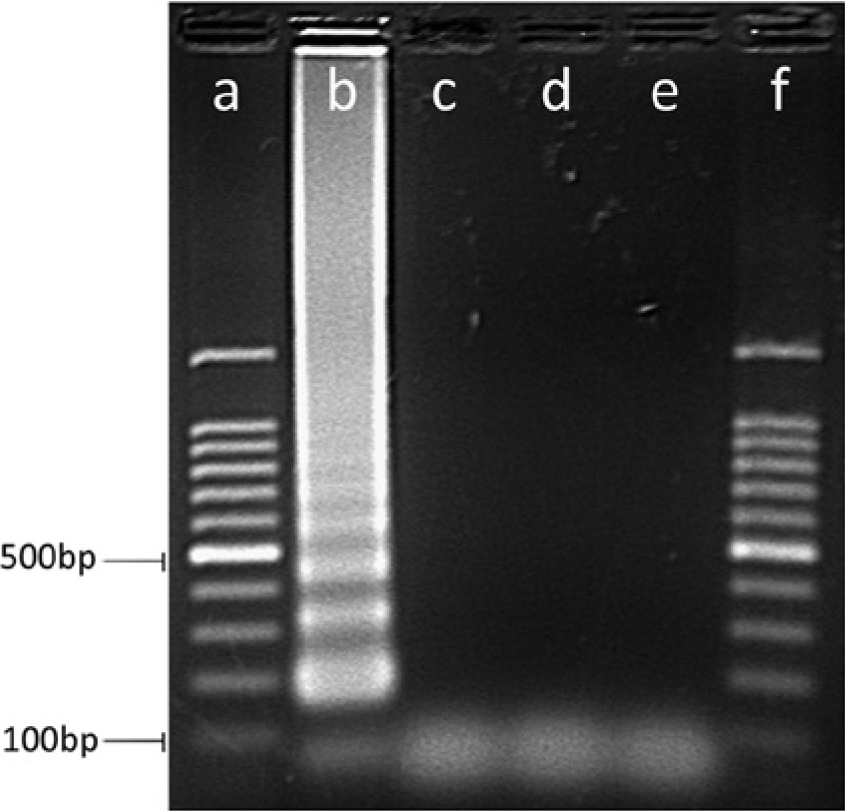
Agarose gel electrophoresis of the spsL loop-mediated isothermal amplification reaction amplicons. Lanes a and f: 100-bp molecular marker; lane b: Staphylococcus pseudintermedius; lane c: Staphylococcus delphini A; lane d: S. delphini B; lane e: no-template control. Amplification was only observed in the reaction containing DNA extract from S. pseudintermedius.
Diagnostic and analytical assay specificity
An initial query of the theoretical amplicon against the NCBI database revealed no cross reaction with other organisms. Subsequent testing using set D DNA extracts (30 different bacterial species), which included the members of the SIG, showed no cross reactions. All set A (isolates from UoL) DNA extracts were detected in less than 8 min. Melting curve analysis of these reactions revealed the characteristic target DNA amplification profile, with a single dissociating temperature of 86.5 ± 0.4°C across all 68 DNA extracts tested. The assay was further tested using DNA extract from 40 clinical S. pseudintermedius (set B) isolates, which included methicillin-resistant strains. The LAMP assay detected all 40 in less than 8 min, with melting curve analysis indicating the presence of a single amplicon with a dissociation temperature of 86.4 ± 0.4°C.
In triplicate reactions, the assay correctly amplifiedS. pseudintermedius DNA from set C (direct-from-swab extracts) in 29 of the 35 extracts tested. In at least 2 out of 3 repeats, 32 samples were detected, while 33 samples were detected in at least 1 out of 3 repeats. This resulted in a direct-from-swab testing diagnostic sensitivity of 85.4% (3/3 repeats), 92.1% (2/3 repeats), and 94.6% (1/3 repeats). Melting curve analysis of the amplified products also revealed the presence of a single product with a dissociation temperature of 86.5 ± 0.4°C. The 2 crude DNA extracts from set C that were negative using the LAMP reaction (not detected in any of the 3 repeats) were detected on subsequent analysis using DNA extract prepared from pure culture grown from the swabs (Table 1).
Analytical sensitivity of the spsL LAMP assay
The analytical sensitivity of the assay, determined through analysis of serially diluted target DNA, was found to be in the order of 10 gene copies per reaction (approximately 0.0052 fg/µl). At this concentration, the target was reliably (3/3 repeats) detected in less than 14 min. As few as 3 copies of the target DNA could also be detected (2/3 repeats) by the assay. An inverse relationship was observed between the concentration of the starting template in the reaction mix and the time taken for the reaction to reach and cross the CT.
Analysis of both real-time and LFD-mediated detection of the reaction’s amplicon, using the same serially diluted template, showed congruency in the minimum concentration of template DNA (0.039 fg/µl) reliably and consistently (3/3 repeats) detectable with both detection systems.
Detection of LAMP products using LFD
All 35 DNA extracts from set C previously tested on the real-time assay, were reanalyzed using the labeled primers and the amplification product detected using an LFD. When detected by means of an LFD, a positive spsL LAMP reaction was illustrated by the presence of a band on the test (T-line) and control (C-line) line of the LFD (Fig. 3). Thirty-two of the 35 extracts tested were detected on the LFD. Of the 3 DNA extracts not detected using a LFD, 2 were also not detected with the real-time LAMP assay. However, as with the real-time LAMP assay, the 2 negative samples were subsequently detected by LFD using DNA extract prepared from a pure culture. Analysis of the amplification profile of the single sample detected by real-time, but not LFD, revealed a significantly late CT value, with the reaction showing a linear amplification profile. A single dissociation peak at 86.9°C, characteristic of the spsL-LAMP amplicon, was observed on melting curve analysis of the reaction.

Detection of a positive spsL loop-mediated isothermal amplification assay (LAMP) reaction using a simple lateral flow device (LFD). The LFD on the left shows a negative reaction, the LFD on the right shows a positive reaction. A band on the T-line illustrates the presence of the target material. A band on the T-line was observed only in the LAMP reaction, which contained DNA extract from Staphylococcus pseudintermedius (LFD on the right). The C-line band serves as an internal control confirming the inoculated material has run the length of the LFD column (both LFDs).
Comparison between LAMP assay and PCR-RFLP
Seventeen of 35 set C DNA extracts analyzed were detected using the PCR-RFLP methodology, compared to 33 (29 in 3/3 repeats) detected by the LAMP assay. The diagnostic sensitivity of the PCR-RFLP method from the crude clinical swab extracts was thus 66%, compared to 85.4% (94.6% in 1/3 repeats) achieved with the LAMP assay. All DNA extracts detected by PCR-RFLP method were also detected on both real-time LAMP and LAMP using LFD detection methodology.
Discussion
In the current study, a rapid and accurate LAMP assay for detection of S. pseudintermedius is described. Compared with conventional PCR, LAMP reactions are characteristically rapid and are performed to higher amplification efficiency. This is due to the isothermal nature of LAMP, as it proceeds at the optimum temperature of the polymerase, with no loss of time resulting from changes in the temperature of the reaction system.7,12 The assay reported in the present study progressed significantly quicker compared to previously reported LAMP assays,10,11,18 reaching its endpoint in less than 14 min. Furthermore, the efficiency of the assay, measured by product yield, was consistent with the yield reported for some LAMP assays. A yield of approximately 394 ng per µl of DNA was observed with the spsL assay, compared to 11.1 µg per 25 µl previously reported for a prostate-specific antigen LAMP reaction. 15
Because the spsL gene has only recently (2011) been identified, 1 the prevalence of this gene among S. pseudintermedius is unknown. It was therefore imperative to evaluate the assay against a wide range of S. pseudintermedius strains originating from different dog breeds, and from geographically dissimilar regions. Detection of the spsL gene in all 108 S. pseudintermedius strains tested suggests a high prevalence of this gene among S. pseudintermedius. Moreover, because cell wall–anchored proteins, encoded by spsL, play an important role in the attachment of the bacteria to host extracellular matrix, 1 this target may be conserved in the species.
Analysis of the assay using serially diluted target DNA showed that target DNA in the order of 10 gene copies per reaction (0.0052 fg/µl) could be reliably (3/3 repeats) detected in under 14 min. As a single copy of the spsL gene is present in S. pseudintermedius (NC_017568), the diagnostic relevance of this observation is that as few as 10 bacteria present in a reaction could be detected.
The diagnostic sensitivity of the assay was explored by analysis using DNA extracts prepared directly from clinical swabs (set C). The assay showed diagnostic sensitivity between 85.4% (3/3 repeats) and 94.6% (1/3 repeats). A diagnostic sensitivity of 94.6% corresponds to detection in 33 out of 35 samples tested. The lack of amplification observed in 2 of the 35 swab samples tested could be due to a paucity of the target material (below the analytical sensitivity of the assay) in the DNA extracts prepared from the clinical swabs. This opinion is reinforced by the fact that the 2 negative DNA swab extracts were subsequently detected when tested using pure-culture DNA extracts originating from the clinical swabs. It is worth noting that the swabs were inoculated on at least 4 agar plates for routine identification prior to extraction, and no product was obtained from the same 2 LAMP-negative DNA swab extracts using PCR-RFLP. The LAMP assay in the current study had a superior diagnostic sensitivity compared to the current PCR-RFLP method when DNA extracts prepared directly from swabs taken from surgical infection sites were tested. Loop-mediated isothermal amplification has previously been noted to be particularly robust, not suffering deleterious effects even when used in concert with biological materials that may otherwise inhibit or reduce the efficiency of a standard PCR reaction. 13
Reliable and convenient detection of the amplified product is an essential step in molecular reactions. Some techniques employed for product detection in LAMP reactions, though generally adequate, may not be wholly suitable when considered as a diagnostic tool.4,19 For instance, colometric and turbidimetric determination of a successful LAMP reaction can be subjective when judged on visual observation alone. However, the use of relatively inexpensive LFDs, as illustrated in the current study, can be efficacious as they allow for clear, precise, and unambiguous confirmation of a reaction outcome. All but 1 sample was detected when testing 33 direct-from-swab DNA extracts that were positive with the real-time LAMP assay. Real-time detection, as illustrated by the single LFD-negative sample, may be slightly more sensitive than LFD. The LFD-negative DNA extract, though with a significantly late CT and still at the linear phase of the reaction after 30 min, was nonetheless detected by real-time analysis. However, subsequent comparison of the analytical sensitivity for both real-time LFD LAMP assays, using the same serially diluted template, showed both systems had precisely the same limit of detection. This suggests that the use of the modified primers and LFD do not have deleterious effects on the LAMP reaction.
Despite the fact that an inferior analytical sensitivity has been reported with some non–agarose-based “naked-eye” detection systems,7,12 the LFD technology used in the present study has shown a detection limit equivalent to that of real-time analysis. In comparison, real-time detection allows for relatively quicker confirmation of a positive reaction, while LFD is limited to end point detection. However, this only makes a difference of approximately 15 min. Conversely, the requirement for the presence of both FITC and biotin for LFD detection provides an additional level of specificity to LFD detection over real-time detection. The newly developed LAMP assay is therefore amenable to offering investigators a choice in the detection method employed, depending on their specific requirements.
The emergence of S. pseudintermedius as a clinical challenge in small animal medicine has called for efficient detection tools. The established PCR-RFLP method for identification and discrimination of S. pseudintermedius from other members of the SIG, though highly effective, is not amenable for use within veterinary practices and non-specialized laboratories. The spsL LAMP assay designed and evaluated in the present study has illustrated its potential as a rapid and efficient molecular diagnostic tool, which may prove useful for accurate detection and identification of S. pseudintermedius within veterinary practices.
Footnotes
Acknowledgements
The authors are grateful to Vanessa Schmidt and Nicola Williams from the University of Liverpool, for their kind and generous donation of some of the bacterial isolates used in this work. The authors are also grateful to Charlotte Batcheldor and Lucy Grist from the University of Surrey for their technical support.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Funding for the project was provided by an organization owned by an author of the article.
Funding
Financial support for the research, authorship, and publication of this article was provided by Fitzpatrick Referrals.
a.
LAMP designer, Premier Biosoft International, Palo Alto, CA.
b.
OptiGene Isothermal Master Mix, OptiGene Ltd, Horsham, United Kingdom.
c.
Mx3000p QPCR system (software v 4.1.0, build 389), Agilent Technologies, Stockport, Cheshire, United Kingdom.
d.
PCRD-2, Pocket Diagnostics, York, North Yorkshire, United Kingdom.
f.
Taq master mix kit, Qiagen GmbH, Hilden, Germany.
g.
QIAquick PCR purification kit, Qiagen GmbH, Hilden, Germany.
h.
NanoDrop 1000, Thermo Scientific, Wilmington, DE.