
Abstract
Bluetongue virus (BTV) and Epizootic hemorrhagic disease virus (EHDV) possess similar structural and molecular features, are transmitted by biting midges (genus Culicoides), and cause similar diseases in some susceptible ruminants. Generally, BTV causes subclinical disease in cattle, characterized by a prolonged viremia. EHDV-associated disease in cattle is less prominent; however, it has emerged as a major economic threat to the white-tailed deer (Odocoileus virginianus) industry in many areas of the United States. The recent emergence of multiple BTV and EHDV serotypes previously undetected in the United States demonstrates the need for robust detection of all known serotypes and differential diagnosis. For this purpose, a streamlined workflow consisting of an automated nucleic acid purification and denaturation method and a multiplex one-step reverse transcription quantitative polymerase chain reaction for the simultaneous detection of BTV serotypes 1–24 and EHDV serotypes 1–7 was developed using previously published BTV and EHDV assays. The denaturation of double-stranded (ds) BTV and EHDV RNA was incorporated into the automated nucleic acid purification process thus eliminating the commonly used separate step of dsRNA denaturation. The performance of this workflow was compared with the World Organization of Animal Health BTV reference laboratory (National Veterinary Services Laboratory, Ames, Iowa) workflow for BTV and EHDV detection, and high agreement was observed. Implementation of the workflow in routine diagnostic testing enables the detection of, and differentiation between, BTV and EHDV, and coinfections in bovine blood and cervine tissues, offering significant benefits in terms of differential disease diagnosis, herd health monitoring, and regulated testing.
Keywords
Introduction
Bluetongue and epizootic hemorrhagic disease are hemorrhagic diseases of ruminants caused by Bluetongue virus (BTV) and Epizootic hemorrhagic disease virus (EHDV), respectively. Bluetongue virus and EHDV (family Reoviridae, subfamily Sedoreovirinae, genus Orbivirus) have segmented genomes composed of 10 segments of double-stranded RNA that encode 7 structural and 4 nonstructural proteins. The segmented viral genomes facilitate reassortment with different serotypes during coinfection with other viruses of the same species.30,46 Serotypes are determined by the viral protein 2 (VP2), an outer capsid protein. To date, 26 serotypes of BTV and 7 serotypes of EHDV have been identified.4,10,20 Until 1998, only BTV serotypes 2, 10, 11, 13, and 17 were present in the United States. Since then, an additional 10 serotypes (1, 3, 5, 6, 9, 12, 14, 19, 22, and 24) have been identified, primarily in the southern areas of the United States. 25 As for EHDV, 2 strains were endemic to the United States (EHDV-1, New Jersey strain, and EHDV-2, Alberta strain).12,34 However, in 2006, an exotic strain of EHDV-6 was isolated from white-tailed deer (Odocoileus virginianus) in Indiana and Illinois. 1 The virus has since been identified throughout the midwestern, eastern, and southern United States.1,2
Both BTV and EHDV are noncontagious viruses spread by biting Culicoides midges. The viruses are endemic in geographical locations with temperate, subtropical, and tropical climates (i.e., areas where the insect vector is present and capable of spreading disease).20,30,46 Wild and domestic ruminants, including sheep, cattle, and white-tailed deer are susceptible to BTV and EHDV. Clinical disease from BTV and/or EHDV infection is most often seen in white-tailed deer and includes fever, excessive salivation and nasal discharge, and hemorrhaging from oral and nasal tissue. 46 Serotypes of BTV found in the United States do not typically cause clinical disease in cattle. 25 However, exotic BTV serotypes are known to cause clinical signs, and occasionally clinical signs are also seen with U.S. serotypes. In late summer through early fall of 2012, a significant number of epizootic hemorrhagic disease cases in cattle were confirmed in the northern U.S. states of Nebraska, South Dakota, Wyoming, and Iowa, and it is believed that the disease was spread from deer to cattle by insect vectors (Wilson D: 2013, Epizootic hemorrhagic disease update. Calif Vet Jan/Feb: 44–45). In general, cattle most often act as reservoirs for BTV and EHDV due to a prolonged cell-associated viremia, which contributes significantly to the epidemiology of the disease. 46
Bluetongue virus and EHDV infections can have a negative economic impact on the cattle and deer industry.19,30,41,46 The emergence of exotic strains of BTV and EHDV, as well as the appearance of EHDV in cattle in northern states, demonstrates the emerging and dynamic nature of these viruses and the potential increasing impact on the economy. Economic impact includes loss of infected animals, trade restrictions imposed on the movement of animals and/or livestock and their products (i.e., semen and ova), and testing costs.19,27,30 Because of the negative economic consequences of BTV and EHDV infection, the World Organization for Animal Health (OIE) has established recommendations for international trade in order to prevent the spread of these viruses to nonaffected countries. 46
Several reverse transcription polymerase chain reaction (RT-PCR) assays, targeting various viral proteins, have been developed for the detection of BTV and EHDV.5,17,43,44 Because the viral genomes are composed of double-stranded RNA (dsRNA), a denaturation step is included prior to nucleic acid amplification. There are currently several methods in use for denaturation of the dsRNA genome, including heat denaturation at 95°C (for 1–8 min) with or without dimethyl sulfoxide15,40,41 and the use of various chemicals such as methylmercury (II) hydroxide,3,45 trehalose,11,13,37 tetramethylene sulfoxide,11,13 and betaine.13,36 While these methods are successful at enhancing amplification of BTV and EHDV, they are labor intensive and create the potential for contamination, as they require an additional step following nucleic acid purification and prior to PCR amplification. In addition, several of the methods require the use of hazardous chemicals.
Therefore, an innovative streamlined workflow that incorporates the denaturation of the viral dsRNA into the nucleic acid purification was developed in order to eliminate the separate step of dsRNA denaturation commonly used for enhanced PCR sensitivity. The workflow consisted of an automated nucleic acid purification and denaturation method and a BTV and EHDV multiplex one-step quantitative RT-PCR (mRT-qPCR) for the simultaneous detection of BTV serotypes 1–24 (BTV serotypes 25–26 were not tested with this workflow because these viruses were unavailable) and EHDV serotypes 1–7. The performance of this workflow was assessed by comparison with the OIE BTV reference laboratory (http://www.oie.int/; the National Veterinary Services Laboratory [NVSL; Ames, Iowa]) workflow for detection of BTV and EHDV. Following successful performance evaluation, the workflow was implemented at the Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL; College Station, Texas) for diagnostic testing of greater than 3,000 samples during a 1-year testing period.
Methods and materials
Virus reference strains
Viral stocks or BTV RNA of serotypes 1–24 were obtained from the NVSL. Epizootic hemorrhagic disease virus serotypes 1 and 2 were obtained from the NVSL, while RNA for EHDV serotypes 3–7 was kindly provided by the Arthropod-Borne Animal Diseases Research Unit, Center for Grain and Animal Health Research, Agricultural Research Service, U.S. Department of Agriculture (Manhattan, Kansas).
Samples for performance evaluation
For the automated nucleic acid purification and RNA denaturation evaluation, the following samples were used: 20 BTV-only positive bovine blood samples, 37 (14 blood and 23 tissue) EHDV-only positive samples, and 33 (30 blood and 3 tissue) BTV and EHDV–positive samples; positive samples were defined by mRT-qPCR. For TVMDL workflow diagnostic sensitivity and specificity determination using the NVSL workflow as the reference method, a total of 125 bovine blood samples (consisting of BTV, EHDV, and BTV and EHDV positives and negatives) were used. Additionally, 126 BTV and EHDV–negative blood samples from the presumed BTV and EHDV–free state of Wisconsin (confirmed by PCR by the Wisconsin Veterinary Diagnostic Laboratory [WVDL; Madison, Wisconsin]) were used to verify assay specificity.
TVMDL workflow
An in vitro transcribed BTV and EHDV control RNA, containing the target sequences for BTV and EHDV, and an in vitro transcribed exogenous internal positive control (XIPC) RNA were generated for mRT-qPCR optimization. DNA sequences specific to BTV and EHDV, as well as XIPC, were chemically synthesized and cloned into a plasmid vector by a commercial company. a Plasmids were linearized using HindIII restriction enzyme digestion and subsequently transcribed into RNA using a commercial transcription kit. b In vitro transcription was performed according to the manufacturer’s instructions. In vitro transcripts were quantitated using a spectrophotometer, c and sizes were verified by electrophoresis. d
The TVMDL oligonucleotides sequence information and the final reaction concentrations in the 25-µl mRT-qPCR are provided in Table 1. Primer and probe sequences for detection of BTV, EHDV, and XIPC were adopted from previous publications.14,16,31 The synthetic construct clone NISTag38 external RNA control sequence (GenBank accession no. DQ883679) is a unique artificial antigenomic sequence with no significant homology to annotated species sequences. This sequence was used as the XIPC target. All primers and probes were purchased from a commercial source. e
Primers and probes for the Bluetongue virus and Epizootic hemorrhagic disease virus multiplex reverse transcription polymerase chain reaction assays.*

FAM = 6-carboxyfluorescein; BHQ1, BHQ2 = black hole quencher 1 and 2, respectively; MGB = minor groove binder; NSP = nonstructural protein; NA = not applicable.
Nucleic acid was purified from all samples at TVMDL using a commercial RNA isolation kit. b Briefly, a 50-µl blood sample, or a 50-µl tissue homogenate (10% weight/volume in 1× phosphate buffered saline), was transferred to a 96-well, deep-well plate containing 20 µl of magnetic bead mix (10 µl of lysis/binding enhancer and 10 µl of RNA binding beads), and 400 µl of lysis binding solution (containing 200 µl of lysis binding concentrate, 1 µl of carrier RNA [1 µg/µl], 1 µl of XIPC RNA [at 10,000 copies/µl] and 200 µl of 100% isopropanol) was added. This plate was denoted as the sample plate. The following four 96-well plates were loaded onto an automatic particle processor c for nucleic acid purification: sample plate, wash solution 1 plate (300 µl/well), wash solution 2 plate (300 µl/well), and elution buffer plate (90 µl/well). The nucleic acid purification procedure consisted of the following steps: lysis/binding for 5 min, one 60-sec wash 1, one 15-sec wash 2, 1-min dry step, and a 3-min heated elution step at 70°C or 95°C. For those samples that were eluted at 70°C, all liquid was transferred to a PCR plate, and then denatured at 95°C for 5 min on a thermal cycler f immediately following elution. All samples were immediately placed on ice following denaturation.
The TVMDL mRT-qPCR utilized the components of a commercial RT-PCR kit b (according to manufacturer’s instructions) and the primers and probes for detection of BTV, EHDV, and the XIPC; the final concentration of each oligonucleotide is provided in Table 1. Each reaction contained 12.5 µl of 2× multiplex RT-PCR buffer, 2.5 µl of 10× multiplex enzyme mix, 1 µl of 25× primer probe mix (containing all the oligonucleotides in Table 1), 1 µl of nuclease-free water, and 8 µl of nucleic acid template for a total volume of 25 µl. The RT-qPCR was performed using a real-time PCR system. b The cycling conditions were as follows: reverse transcription at 48°C for 10 min (single cycle), activation/denaturation at 95°C for 10 min (single cycle), and 40 cycles of amplification at 95°C for 15 sec and 55°C for 45 sec. For diagnostic sensitivity and specificity determination using the NVSL workflow as the reference method, samples with a quantification cycle (Cq) ≤36.0 cycles were considered positive for BTV, while samples with a Cq ≤40.0 cycles were considered positive for EHDV.
NVSL (OIE BTV reference laboratory) workflow
The sequences of the oligonucleotides used in the nested PCR (nPCR) are provided in Table 2. Primer sequences for detection of BTV and EHDV were obtained from previous publications.18,42 Nucleic acid was purified at the NVSL using a commercial reagent per the manufacturer’s instructions. g Each purified nucleic acid sample was resuspended in 25 µl of RNase-free water, 5 µl was transferred to a PCR tube, and the samples were heated at 95°C for 10 min on a thermal cycler b to denature the dsRNA. Samples were cooled at 4°C prior to use in the RT-PCR.
Primers for the Bluetongue virus and Epizootic hemorrhagic disease virus nested reverse transcription polymerase chain assays.*
NSP = nonstructural protein.
The final concentrations (per reaction) of the oligonucleotides for BTV and EHDV in the RT-PCR and secondary nPCR are provided in Table 2. Each RT-PCR reaction contained 27.0 µl of RNase-free water, 5.0 µl of 10× PCR buffer (100 mM Tris–HCl, pH 8.3, 500 mM KCl), b 4.0 µl of 25 mM MgCl2, b 4.0 µl of 10 mM deoxyribonucleotide triphosphate (dNTP) mix, b 0.4 µl of RT enzyme (200 units/µl), g 0.125 µl of RNase inhibitor (10 units/µl), g 0.5 µl of DNA polymerase (5 units/µl), b and 4.0 µl of outer primers A and B (30–40 pmol of each primer for BTV or EHDV). Denatured template (5 µl) was mixed with 45 µl of the RT-PCR mixture and amplified using a thermocycler. f The RT-PCR cycling conditions were as follows: 44°C for 50 min, 95°C for 10 min, 35 cycles of amplification at 95°C for 25 sec, 60°C for 20 sec, and 72°C for 25 sec, with a final 5-min extension at 72°C, followed by refrigeration. The secondary PCR reaction (nPCR) contained 32.0 µl of RNase-free water, 5.0 µl of 10× PCR buffer (100 mM Tris–HCl, pH 8.3, 500 mM KCl), b 3.5 µl of 25 mM MgCl2, b 4.5 µl of 10 mM dNTP mix, b 0.5 µl of DNA polymerase (5 units/µl), b and 4.0 µl of nested primers C and D (20–30 pmol of each primer for BTV or 30–40 pmol of each primer for EHDV). The initial RT-PCR amplicon (1.5 µl) was mixed with 48.5 µl of the nPCR mixture. The cycling conditions for the nPCR were as follows: 95°C for 10 min (single cycle), 35 cycles of amplification at 95°C for 20 sec, 60°C for 20 sec, 72°C for 20 sec, with a final 5 min hold at 72°C, followed by refrigeration.
Amplified PCR products were visualized on a 2.5–3% agarose gel. h Amplified DNA products (8–10 µl) were mixed with 3 µl of DNA loading buffer containing a DNA stain, i loaded onto the gel, and run at 65–80 V for 1–1.5 hr. Bands were visualized under ultraviolet light. All samples were tested in duplicate, and only those samples containing bands of the correct size (101 bp for BTV and 138 bp for EHDV) for both duplicates were considered positive.
Statistical analysis
Regression analysis was performed using statistical software j to assess correlation between the automated RNA denaturation and thermal cycler denaturation. Probit analysis was performed using statistical software j to assess the limit of detection of each assay.8,9,35 Repeatability of the mRT-qPCR was determined by calculation of the intra- and interassay coefficient of variation (CV) for each pathogen assay. Intra-assay variability was analyzed in triplicate PCR at each dilution, and interassay variability was analyzed in 3 separate experiments (runs). Intra-assay CV = [(average of each run’s Cq standard deviation (SD)/average of each run’s Cq average) × 100]; interassay CV = [(SD of the averages of each run/average of the averages of each run) × 100].
Performance of the TVMDL workflow for the detection of BTV and EHDV was evaluated by comparison against the NVSL method using a panel of 125 positive and negative blood samples. Cohen’s kappa and percentage agreement were calculated using a previously published method. 24
Results
Automated 95°C denaturation versus separate thermal cycler 95°C denaturation
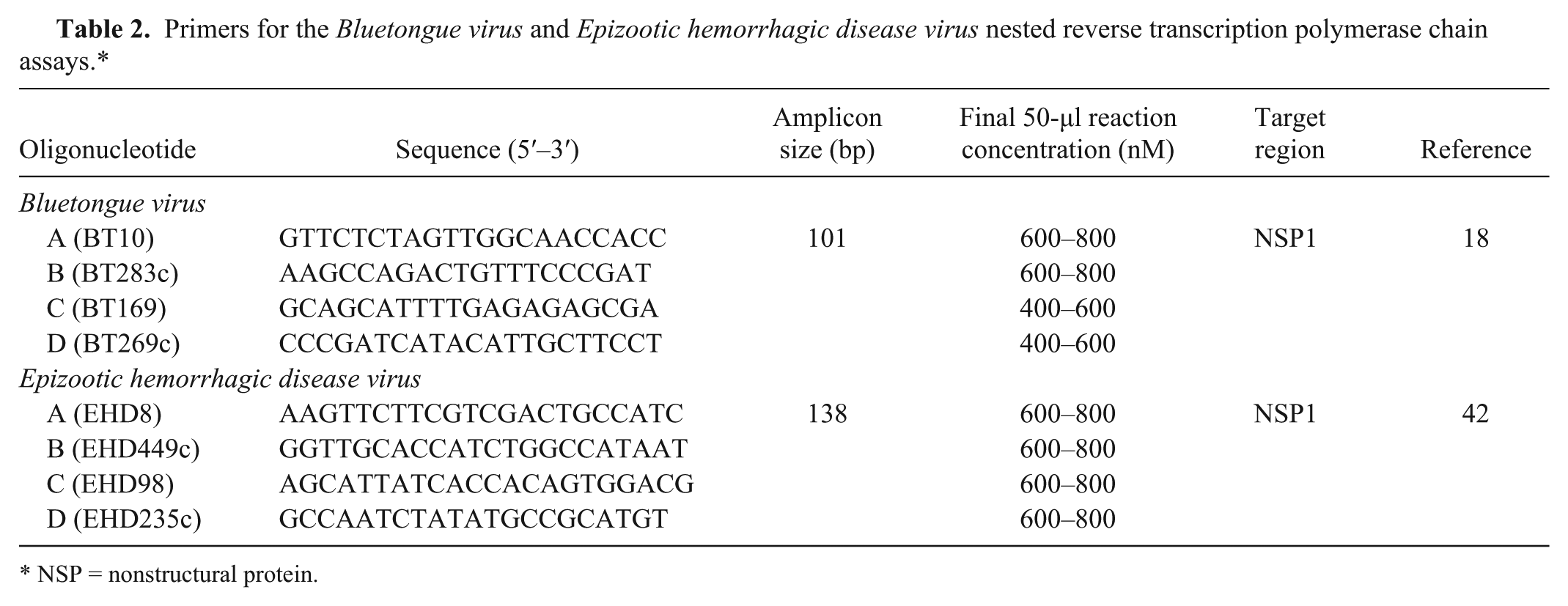
The performance of the automated 95°C denaturation was compared to the separate thermal cycler 95°C denaturation using 90 positive samples consisting of 20 BTV-only positive, 37 EHDV-only positive, and 33 BTV and EHDV–positive samples. The automated 95°C denaturation produced equivalent or lower Cq values to the thermal cycler denaturation (Supplemental Fig. 1A, 1B). The XY scatter plots of thermal cycler denaturation Cq values versus the automated denaturation Cq values showed a good correlation between the 2 denaturation methods (Fig. 1A, 1B). Correlation coefficients for BTV and EHDV were 0.9655 and 0.9438 (P values <0.0001 for both), respectively, indicating that the 2 methods produced equivalent Cq values.
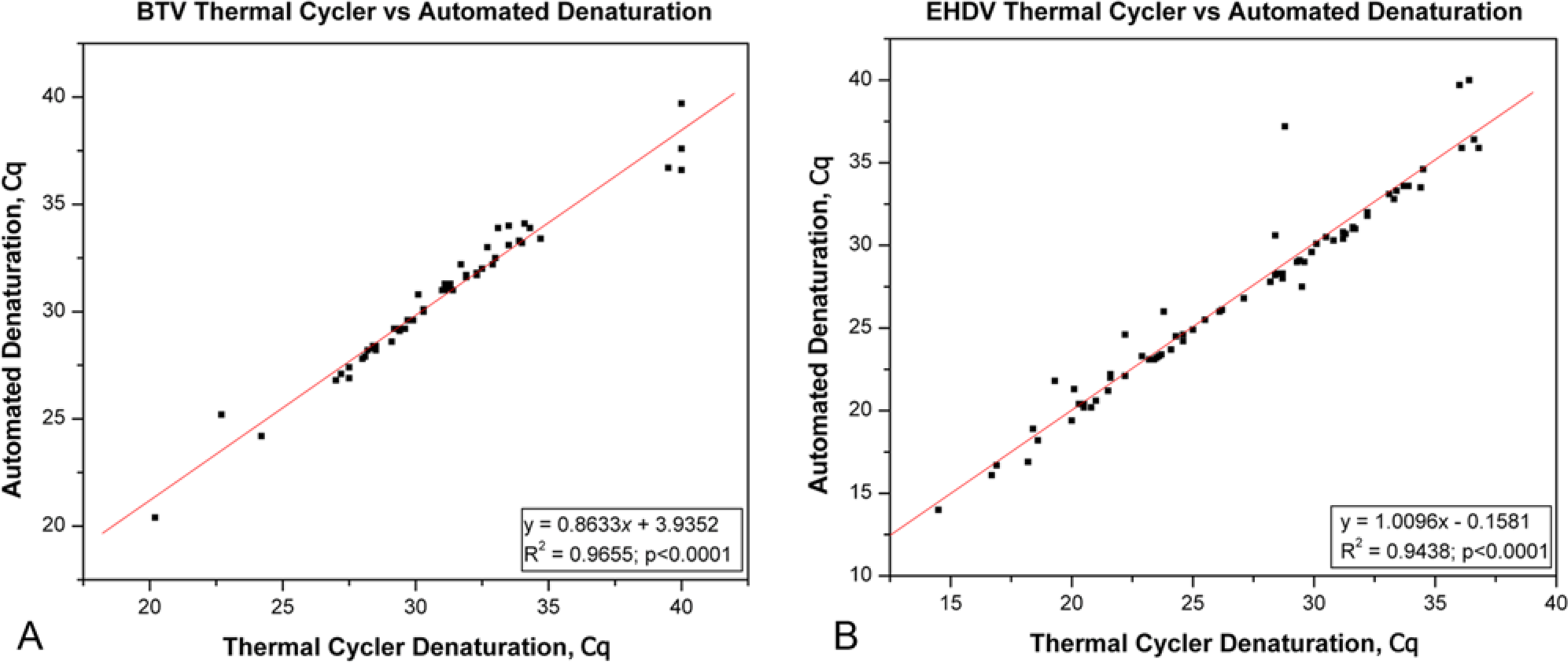
Correlation of the automated 95°C denaturation versus the separate thermal cycler 95°C denaturation of Bluetongue virus (BTV) and Epizootic hemorrhagic disease virus (EHDV) purified RNA. The XY scatter plots of the quantification cycle (Cq) values for the thermal cycler denaturation versus the Cq values for the automated denaturation showed a good correlation between the 2 methods.
Intra- and interassay repeatability and analytical sensitivity of BTV and EHDV assays
Triplicate mRT-qPCR assays in 3 separate experiments were performed in order to assess the intra- and interassay repeatability for the TVMDL BTV and EHDV assays as well as the analytical sensitivity using probit analysis. The CV within runs (intra-assay variability) ranged from 0.4% to 5.3% for BTV and 0.4% to 2.2% for EHDV; between runs (interassay variability) ranged from 1.1% to 5.4% for BTV and 0.5% to 1.8% for EHDV (Supplemental Table 1). Serial dilutions of an in vitro transcribed BTV and EHDV control RNA and XIPC RNA (1,000 copies/reaction) were used to estimate the analytical sensitivity of each assay. Probit analysis was performed for each assay in order to determine the 95% mRT-qPCR detection rate. The data sets for the last 5 purified nucleic acid dilutions of the intra- and interassay variability experiment were used for the analysis. A total of 9 reactions (responses) were used for each BTV- and EHDV-purified nucleic acid dilution; Cq values below 40 were considered a positive response or amplification. Copy number equivalents for the pathogen-purified nucleic acid dilutions were estimated using the mRT-qPCR targeting serial dilutions of BTV and EHDV control RNA and subsequent standard curve data analysis. The mRT-qPCR detection limit was less than 200 BTV target copies and less than 100 EHDV target copies per PCR for both assays, as determined by probit analysis. The probit graph (probability vs. log10 [dose] EHDV-purified nucleic acid) for EHDV is displayed in Figure 2 (similar results were obtained for BTV, data not shown).
Probit analysis of Epizootic hemorrhagic disease virus (EHDV) detection by the multiplex reverse transcription polymerase chain reaction assay. Serial dilutions of EHDV purified nucleic acid were prepared in pathogen-negative bovine blood nucleic acid and used for probit analysis. A total number of 9 responses for each nucleic acid dilution were analyzed in order to determine the nucleic acid dilution corresponding to a detection rate of 95%. Similar results were obtained for Bluetongue virus.
Analysis of diagnostic samples: comparison of NVSL and TVMDL workflows
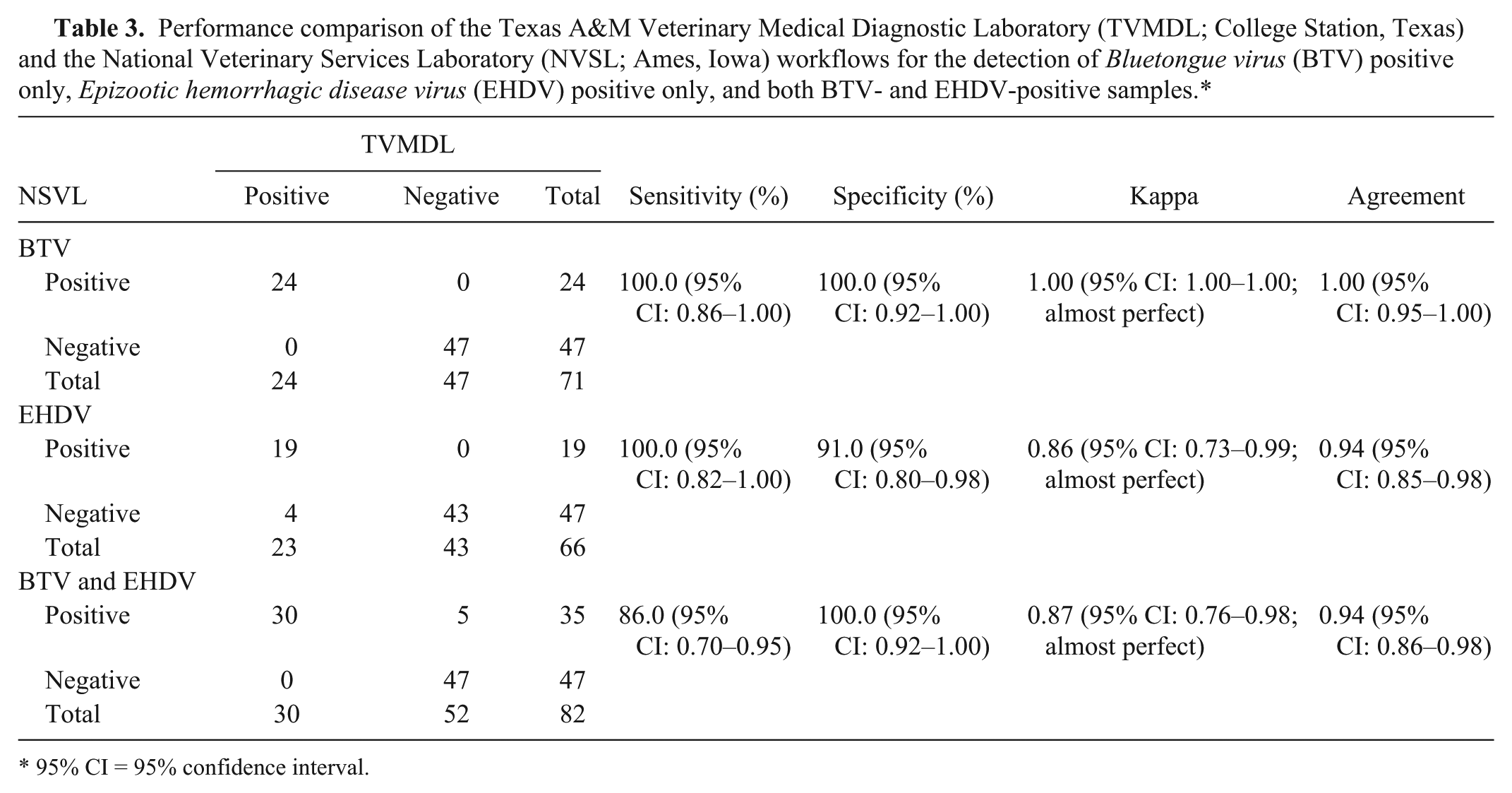
The performance of the TVMDL workflow was evaluated for the detection of BTV and EHDV using a selection of 125 bovine blood samples submitted to the TVMDL. The samples were tested by the TVMDL and NVSL workflows. The NVSL (OIE BTV reference laboratory) workflow was selected as the reference method for diagnostic sensitivity and specificity analysis (Table 3). The NVSL workflow identified 24 (19%) BTV-only positives, 19 (15%) EHDV-only positives, 35 (28%) both BTV and EHDV positives, and 47 (38%) negatives (both BTV and EHDV).
Performance comparison of the Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL; College Station, Texas) and the National Veterinary Services Laboratory (NVSL; Ames, Iowa) workflows for the detection of Bluetongue virus (BTV) positive only, Epizootic hemorrhagic disease virus (EHDV) positive only, and both BTV- and EHDV-positive samples.*
95% CI = 95% confidence interval.
The TVMDL workflow produced equivalent and reliable results. The internal positive control, XIPC RNA, was detected in all samples (average Cq = 31.8 ± 0.5), indicating effective nucleic acid purification and amplification. For the 24 NVSL workflow BTV-only positives, the TVMDL workflow identified all 24 BTV positives (perfect agreement); 21 were identified as BTV-only positive and 3 as both BTV and EHDV positive (EHDV Cq values = 33.5, 34.6, 38.8). For the 19 NVSL workflow EHDV-only positives, the TVMDL workflow identified all 19 positives (perfect agreement). For the 35 NVSL workflow samples that were both BTV and EHDV positive, the TVMDL workflow identified 30 as both BTV and EHDV positive, 2 as BTV-only positive (with no EHDV amplification, Cq values = 0), and 3 as EHDV-only positive. For the 3 EHDV-only positives, BTV amplification was positive with Cq values > 36.0 (Cq values = 39.7, 37.6, 37.2), which was greater than the Cq cutoff value established for identification as BTV positive as determined based on assay repeatability analysis. Thus, these samples were identified as BTV negative.
Lastly, for the 47 NVSL workflow negatives (both BTV and EHDV), the TVMDL workflow identified 4 as EHDV-only positive (Cq values = 31.8, 32.4, 35.1, 37.0). The TVMDL workflow agreement with the NVSL workflow defined “positive” for BTV-only positives was 100% (κ = 1.00, almost perfect), for EHDV-only positives was 94% (κ = 0.86, almost perfect), and both BTV and EHDV positives was 94% (κ = 0.87, almost perfect; Table 3). The agreement for identification of all BTV positives and all EHDV positives, irrespective of single pathogen or dual pathogen positive classification, was calculated to be 98% (κ = 0.95, almost perfect) and 93% (κ = 0.85, almost perfect), respectively (Table 4). This same sample panel was also tested with singleplex BTV or EHDV RT-qPCR, and comparable results with mRT-qPCR were obtained (data not shown) supporting the use of the mRT-qPCR assay. For additional workflow specificity assessment, 126 BTV and EHDV–negative blood samples from the presumed BTV and EHDV–free state of Wisconsin (samples were confirmed negative by PCR by WVDL) were tested and confirmed to be negative using the TVMDL workflow.
Performance comparison of the Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL; College Station, Texas) and the National Veterinary Services Laboratory (NVSL; Ames, Iowa) workflows for the detection of all Bluetongue virus (BTV)–positive samples and all Epizootic hemorrhagic disease virus (EHDV)–positive samples, irrespective of single pathogen or dual pathogen positive classification.*
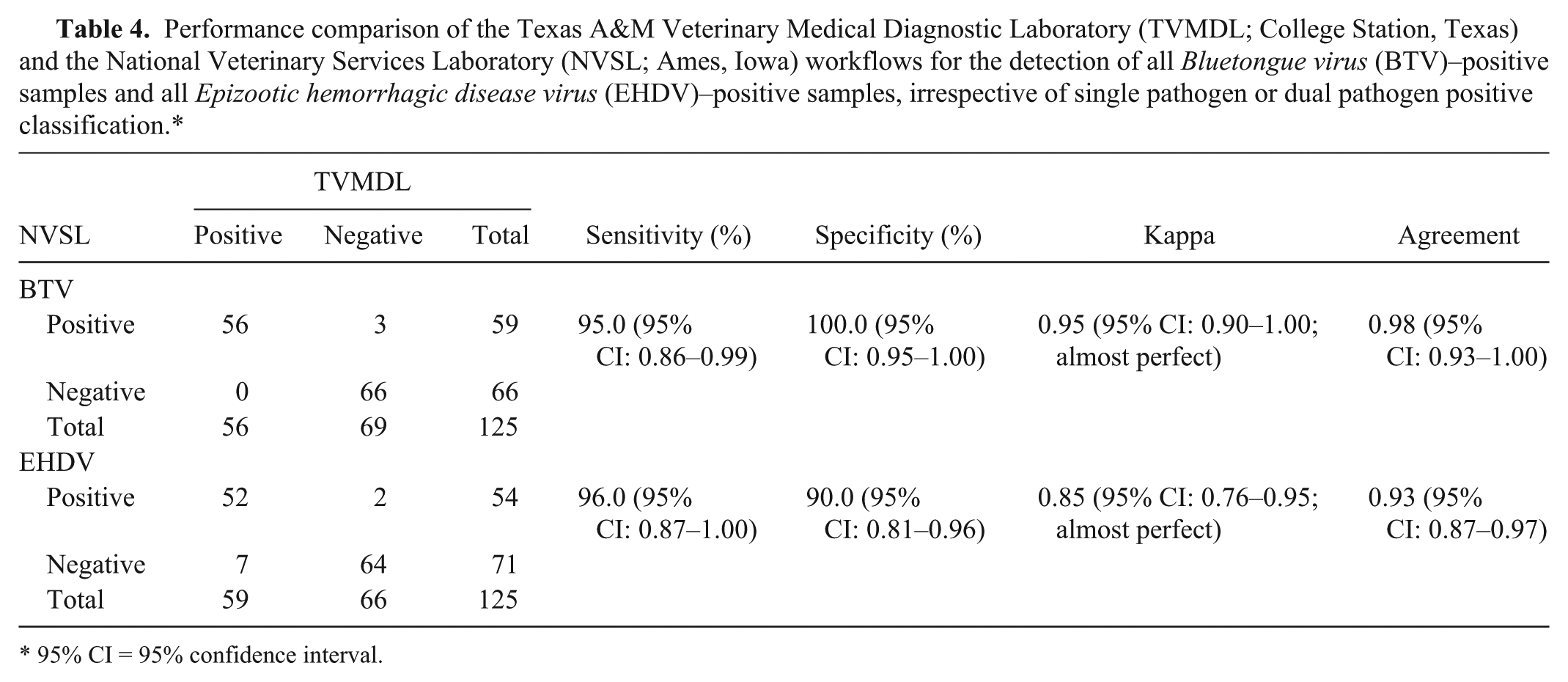
95% CI = 95% confidence interval.
Analysis of TVMDL diagnostic samples
On completion of the successful performance evaluation study, the TVMDL workflow was implemented for routine testing of diagnostic samples submitted to the laboratory for regulatory requirements, herd health management, and clinical diagnosis. Over the course of 1 year (December 2011–December 2012), approximately 3,043 samples consisting of bovine, ovine, caprine, and cervine specimens (i.e., blood, tissue, and semen) were tested as diagnostic submissions (Table 5). A total of 376 positives (approximately 12.4% positive rate) were identified; specifically, 157 BTV-only positives (5.2% positive rate), 176 EHDV-only positives (5.8%), and 43 both BTV and EHDV positives (1.2%). Bluetongue virus and EHDV coinfections were observed at an 11.4% rate (of all positives, 43/376).
Bluetongue virus (BTV) and Epizootic hemorrhagic disease virus (EHDV) diagnostic submissions and outcomes over a 1-year period (n = 3,043 samples total).
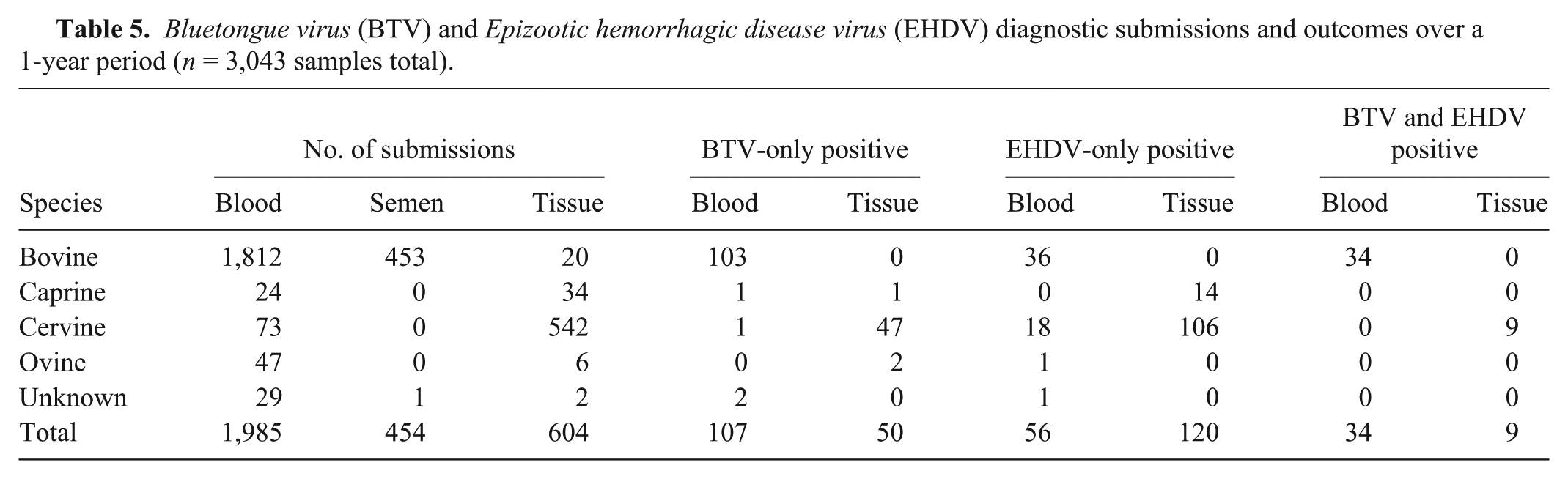
Of the 157 BTV-only positives, 103 were from bovine blood, 48 were from cervine tissues, and the remaining 6 were from ovine, caprine, or unknown sources; the cervine positive tissues consisted of mainly spleen (32/48, approximately 67%), while other tissues such as tonsil, lung, testes, and lymph node represented a minor set of positive tissue samples. Limited clinical hemorrhagic disease information was provided in the submissions thus correlation of infection with disease was not possible. However, it is interesting to note that of the BTV-only positive samples, 2 bovine blood samples and 37 cervine tissue samples (approximately 77% of 48 BTV-only cervine tissue positives) were from animals with clinical hemorrhagic disease; the remaining 112 samples (101 bovine and 11 cervine) lacked clinical disease information.
Of the 176 EHDV-only positives, 124 were from cervine blood (n = 18) and tissue (n = 106), 36 were from bovine blood, and the remaining 16 were from caprine tissue (n = 14), ovine (n = 1), or unknown sources (n = 1). The majority of positive tissue samples (96/120, 80%) were spleen, similar to the BTV positive tissues. All 36 bovine samples and 41 cervine samples lacked clinical disease information. However, 83 of the 124 total cervine EHDV-only positives (approximately 67%) were from animals with clinical disease
Interestingly, of the 43 both BTV and EHDV–positive samples identified, 34 were from bovine blood samples. Two of these 34 coinfected bovine samples were from animals with clinical signs; both animals were from farms reporting a BTV and/or EHDV outbreak; the remaining 32 samples lacked disease information. The other 9 BTV and EHDV copositive samples were from cervine tissues (spleen, testes, tonsil, and salivary gland), 5 of which were from animals with disease, while 4 were samples submitted without disease information. Neither BTV nor EHDV was detected in any of the semen (n = 454) samples submitted for diagnostic testing.
Discussion
Bluetongue virus and EHDV are 2 viral diseases of economic importance to the cattle and white-tailed deer industry in the United States.19,41,46 Identification of these viruses is important in order to evaluate their distribution and epidemiology. The current available reference laboratory PCR method for the detection of BTV and EHDV involves 2 separate nPCR reactions, 1 for each virus. 46 Additionally, in order to enhance sensitivity, the purified double-stranded viral RNA is denatured in a separate step following nucleic acid purification and prior to PCR amplification.13,27,40 The dsRNA denaturation step, however, is time consuming, requires the use of hazardous chemicals, and can create the potential for contamination and human error due to additional pipetting steps. In order to reduce potential errors and possible contamination from these additional pipetting steps, an innovative streamlined workflow was developed for the rapid denaturation and subsequent detection of BTV (serotypes 1–24) and EHDV (serotypes 1–7). Denaturation (95°C for 3 min) of the viral dsRNA was incorporated into the elution step of the automated particle processor c program. Therefore, the viral nucleic acid was denatured during nucleic acid purification, thus eliminating the need for a separate denaturation step prior to RT-qPCR amplification.
Comparison of the automated 95°C denaturation method and the commonly used separate thermal cycler 95°C denaturation step method demonstrated that the automated denaturation method produced equivalent or lower Cq results for the detection of BTV and EHDV. Correlation coefficients of 0.9655 and 0.9438 (P values <0.0001 for both) for BTV and EHDV, respectively, revealed a strong correlation between both denaturation methods.
The streamlined workflow presented herein, consisting of automated denaturation and purification followed by BTV and EHDV mRT-qPCR amplification, was assessed against the thermal cycler denaturation and nPCR workflow performed at the NVSL, an OIE BTV reference laboratory. When compared with the NVSL workflow, the TVMDL workflow showed a diagnostic sensitivity of 95.0% for BTV positives and 96% for EHDV positives, and a diagnostic specificity of 100.0% for BTV-positive and 90.0% for EHDV-positive samples, respectively, irrespective of single pathogen or dual pathogen positive classification. Overall, the results demonstrate very good agreement between the 2 workflows and greater than 95% sensitivity for the TVMDL workflow, thus providing support for the applied utility of this method for BTV and EHDV detection. Practical advantages of the TVMDL workflow include dsRNA denaturation during the automated nucleic acid purification elution step, inclusion of an internal positive control for nucleic acid purification and detection, and concurrent detection and analysis of BTV and EHDV in a single reaction, which reduces testing costs. In addition, inclusion of the internal control reduces the potential of reporting false-negative results due to system and human error, thus providing more confidence in the results. The described automated nucleic acid purification method is also the universal pathogen nucleic acid purification method that is used for diverse biological samples that are submitted for testing at TVMDL, thus enabling sample batching and minimizing labor, equipment, and consumables usage.
The utility of this workflow was demonstrated in the routine diagnostic testing of approximately 3,043 samples submitted to TVMDL during a 1-year period. Of the 376 positive samples identified, 41% (157/376) were BTV-only positives, 47% (176/376) were EHDV-only positives, and, interestingly, 11.4% (43/376) were both BTV and EHDV–positive coinfections.
There have been several reports in the literature of natural BTV and EHDV coinfections, multiple BTV serotype coinfections, and BTV or EHDV reassortants in both cattle and sheep,28,39,42 as well as in the insect vector. 29 Multiple reports from India have detected BTV reassortments in sheep between BTV-16 and BTV-21, 32 BTV-3 eastern and western, 21 BTV-2 eastern and western, 22 and BTV-23 eastern and western topotypes. 23 In the United States, a BTV reassortant that included genes from BTV-2 and BTV-6 was reported in a cow with BTV clinical disease. 26 Epizootic hemorrhagic disease virus reassortants, such as an EHDV-6 (Indiana) that contained genes from endemic EHDV-2 (Alberta) and exotic EHDV-6 (Australia) serotypes, was first recovered from dead white-tailed deer in Indiana and Illinois in 2006. 1 In addition to reassortants, dual BTV serotype coinfections have also been reported: BTV-1 and BTV-12, BTV-3 and BTV-24, and BTV-22 and BTV-24, in several cattle with no clinical signs of disease in Kenya, 39 and BTV-4 and BTV-16, and BTV-4 and BTV-24 in 2 different cows, respectively, with BTV clinical signs in Israel. 7 Because several of these reports identified multiple BTV serotype coinfections in animals with and without clinical signs, the significance of BTV serotype coinfections in disease manifestation is currently unclear. Other factors such as breed and age may play a role in disease presentation. Several of the reports7,39,46 do, however, speculate that these reassortants may be more virulent than the parental serotypes and may therefore be responsible for the increased virulence observed in diseased animals. However, additional studies are needed to verify this assumption.
With regard to BTV and EHDV coinfections, an investigation of the Reunion Island cattle population consisting of 116 cows exhibiting BTV and EHDV clinical signs reported a 4% (5/116 total blood samples tested) BTV-2–EHDV-6 coinfection rate. 28 Because 91% of this cattle population were EHDV-6–only positive, and the coinfected cattle were infected with BTV-2, which, according to the authors, has never caused clinical signs, the authors speculate that the clinical signs displayed in the BTV-2 and EHDV-6 coinfected cattle may be a result of EHDV-6. In this report, 28 BTV-only infections were not detected. Another report identified a 58.6% coinfection rate (58/99 total cow blood samples tested) in nondiseased cows in Kenya. 39 In the current study, BTV and EHDV coinfections were identified in cattle at a rate of 1.49% (34/2,285 total bovine samples tested), and in white-tailed deer (cervine) at a rate of 1.46% (9/615 total cervine samples tested). Two of the 34 coinfected cattle exhibited clinical disease and were from farms reporting a BTV and/or EHDV outbreak; 5 of 9 coinfected white-tailed deer exhibited clinical disease; and the remaining coinfected samples lacked disease information, thus disease correlation with these coinfections is unknown. Based on the available published literature, the current study has evaluated the highest number of animals for BTV and EHDV coinfections thus far in a U.S. sampled population. The importance of these coinfections in the transmission and virulence of these viruses is currently unknown, as there are neither reports of reassortment events between BTV and EHDV, nor reports of exacerbated clinical disease with coinfections. Nevertheless, these coinfections may be important in understanding disease presentations. Similarly, coinfections among multiple BTV serotypes or multiple EHDV serotypes may be important in virus transmission as they create opportunities for potential interaction between several BTV or EHDV serotypes that can potentially result in the generation of newer, more virulent BTV or EHDV serotypes.6,33,38 The potential emergence of new virulent serotypes or the invasion of serotypes previously unseen in certain geographic locations therefore emphasizes the need for rapid identification and differentiation of these 2 viruses that present similar clinical signs.
In summary, a streamlined workflow that includes an innovative automated dsRNA denaturation during nucleic acid purification, followed by the simultaneous detection of BTV and EHDV by mRT-qPCR, has been developed. This workflow provides rapid, reliable results, and reduces the potential for contamination and human error by eliminating the separate denaturation step commonly used to denature BTV and EHDV dsRNA. Implementation and ongoing use of this workflow has enabled detection of single and dual infections in diverse samples for regulatory testing, herd health management, and differential disease diagnosis. The simultaneous and differential detection of BTV and EHDV provides significant benefits for 2 important ruminant diseases with similar clinical presentations, particularly in light of reports of BTV- and EHDV-associated clinical disease in cattle.
Footnotes
Acknowledgements
The authors would like to acknowledge the Wisconsin Veterinary Diagnostic Laboratory and the Arthropod-Borne Animal Diseases Research Unit, Center for Grain and Animal Health Research, USDA, Agricultural Research Service for procurement of samples.
a.
pBluescript II SK+ plasmid vectors containing DNA sequences specific to BTV, EHDV, and XIPC, Blue Heron Biotechnology, Bothell, WA.
b.
Primer Express 3.0 software, MEGAscript T7 High Yield Transcription Kit, MagMAX-96 Viral RNA Isolation Kit, MagMAX Lysis/Binding Solution Concentrate, 7500 Fast Real-Time PCR System, GeneAmp PCR System 9700, AmpliTaq Gold DNA polymerase, 10X PCR Buffer, 25 mM MgCl2, 10 mM dNTP mix (contains 2.5 mM dATP, 2.5 mM dTTP, 2.5 mM dCTP, 2.5 mM dGTP); Applied Biosystems, Carlsbad, CA.
c.
Nanodrop spectrophotometer, KingFisher 96 Magnetic Particle Processor; Thermo Fisher Scientific, Wilmington, DE.
d.
QIAxcel, Qiagen Tissue Lyser; Qiagen Inc., Valencia, CA.
e.
Primers and probes, Biosearch Technologies, Novato, CA.
f.
Thermal Cycler, Eppendorf North America, Hauppauge, NY.
g.
Trizol LS, M-MLV reverse transcriptase (200 units/µl), ribonuclease inhibitor (10 units/µl); Invitrogen Corp., Grand Island, NY.
h.
Wide Mini-Sub Cell System, Bio-Rad Laboratories, Hercules, CA.
i.
GelRed nucleic acid stain, Biotium Inc., Hayward, CA.
j.
NCSS 2007 software, NCSS LLC, Kaysville, UT.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.