
Abstract
A new strain of Aeromonas hydrophila has been implicated in significant losses in farm-raised catfish. Outbreaks attributable to this new strain began in Alabama in the summer of 2009 and have spread to Arkansas and Mississippi in subsequent years. These outbreaks mostly afflicted market-sized fish and resulted in considerable losses in short periods of time. The present research was designed to develop an expeditious diagnostic procedure to detect the new strains of A. hydrophila due to the rapid onset and biosecurity concerns associated with this new disease. A discriminatory quantitative polymerase chain reaction assay was developed using gene sequences unique to the virulent strains identified in a related comparative genomic study. Using this assay, suspect colonies on a culture plate can be positively identified as the new strain within 2 hr. The assay is repeatable and reproducible with a linear dynamic range covering 8 orders of magnitude and a sensitivity of approximately 7 copies of target DNA in a 15-µl reaction. In addition, the assay is able to detect and quantify the virulent strain from catfish tissues (0.025 g), pond water (40 ml), and sediments (0.25 g) with a sensitivity limit of approximately 100 bacteria in a sample. This assay provides rapid discrimination between the new virulent strain and more common A. hydrophila and is useful for epidemiological studies involving the detection and quantification of the virulent strain in environmental samples and fish tissues.
Introduction
Aeromonas hydrophila is a Gram-negative motile bacillus that is ubiquitous in aquatic environments, often presenting as a secondary infection associated with other diseases in cultured catfish. 7 Recently, a highly virulent strain of A. hydrophila has emerged and appears to be acting as a primary pathogen in market-sized catfish. Disease outbreaks attributable to this new strain began in Alabama in the late summer of 2009 and have spread to Arkansas and Mississippi in subsequent years. 9
Diagnosticians assisting catfish farms with motile aeromonad septicemia cases base critical treatment and biosecurity decisions on the strain of A. hydrophila involved in the outbreak. Opportunistic A. hydrophila strains are often encountered secondary to other infectious agents and are generally not of primary concern. 7 However, with the new virulent A. hydrophila (VAh) strain, the response is likely to involve aggressive treatments and rigorous biosecurity. As such, there is an urgent need for a rapid diagnostic test that can clearly distinguish the new strain from strains more commonly associated with secondary infections.
Quantitative polymerase chain reaction (qPCR) assays have become commonplace for the detection of bacterial, viral, and parasitic pathogens of fish (Getchell RG, Bowser PR: 2011, Real-time PCR assays for fish pathogens. In: Proceedings of the Third Bilateral Conference between Russia and the United States: bridging American and Russia with shared perspectives on aquatic animal health, ed. Cipriano RC, Bruckner A, Shchelkunov IS, pp. 168–175. Khaled bin Sultan Living Oceans Foundation, Landover, Maryland). 10 Furthermore, qPCR has been useful to quantitate microbes from a wide variety of environmental samples.1,6,14 However, the use of molecular methods for pathogen detection in catfish aquaculture is complicated by the presence of compounds within catfish ponds that are inhibitory to the amplification of nucleic acids by PCR.3,4 Heavy metals, humic and phenolic compounds, as well as constituents of bacterial cells have all been identified as potential inhibitors commonly associated with environmental samples. 16 Recently, protocols have been developed for qPCR detection and quantification of pathogens from catfish ponds specifically targeting and removing environmental inhibitors during sample processing and analysis.3,4 The present study describes the development of a quantitative real-time PCR assay, termed the VAh qPCR, that rapidly discriminates between VAh strains and strains that are routinely found in aquatic environments and as normal flora of freshwater fish. Moreover, the effectiveness of the assay for detection and quantification of VAh strains from various sample matrices is demonstrated.
Materials and methods
Bacterial isolates
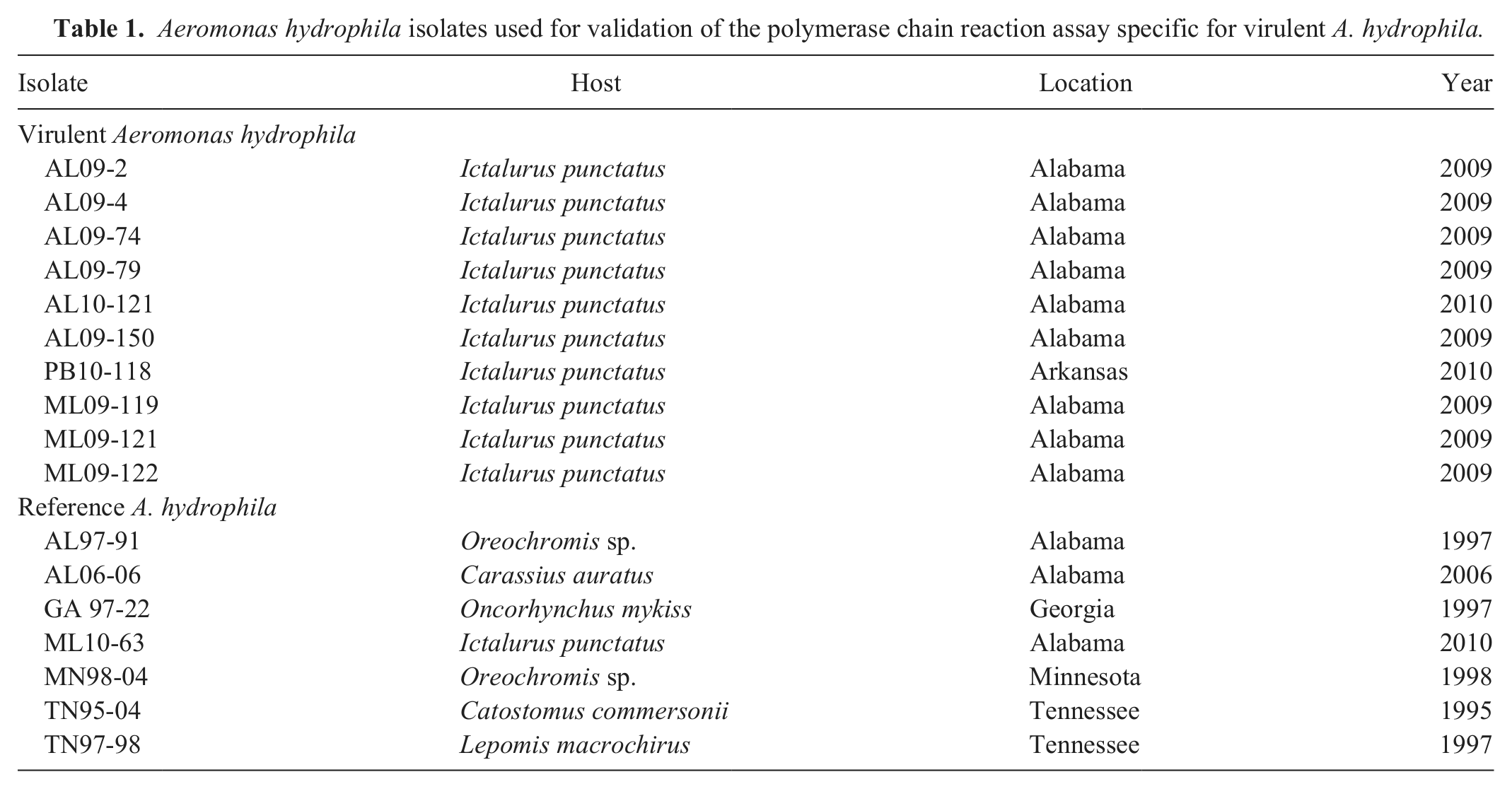
The A. hydrophila strain used in the development of the assay described herein was a case isolate (ML09-121) originally obtained during an outbreak of A. hydrophila from a commercial catfish operation. The isolate was confirmed to be A. hydrophila biochemically and by DNA sequencing of the gyrB gene. Subsequent sequencing of the genome demonstrated this strain was >97% identical to other sequenced VAh strains (Hossain et al., manuscript in preparation). The isolate was grown in brain-heart infusion (BHI) broth a prior to storage in 20% glycerol at −80ºC. Frozen cultures were streaked onto Mueller–Hinton agar a plates supplemented with 5% sheep blood and incubated for 24 hr at 27°C, after which individual colonies were isolated and cultured in 1 ml of BHI broth at 27°C. Along with strain ML09-121, 9 VAh isolates from disease outbreaks in 2009 and 2010 and 7 reference A. hydrophila (RAh) isolates were included in the validation of the assay (Table 1).
Aeromonas hydrophila isolates used for validation of the polymerase chain reaction assay specific for virulent A. hydrophila.
DNA isolation from bacterial cultures
Genomic DNA (gDNA) was isolated from bacterial cultures using a commercial kit, b following the manufacturer suggested protocol for Gram-negative bacteria. In addition, an RNase A digestion was included in the procedure to reduce the influence of RNA on spectrophotometric quantification.
Design and optimization of primer and probe sets for the VAh PCR
Sequence targets unique to the VAh strains were identified by comparative genomic analyses between the VAh and RAh strains (Hossain et al., manuscript in preparation). Primer and probe design 11 targeted a 167-bp region of a predicted open reading frame (609 bp) that was unique to the VAh strains and lacked any significant BLASTn or BLASTx hits (E-value < 0.01) within the GenBank nonredundant nucleotide database (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). The probe was 5’-labeled with 6-carboxyfluorescein, an internal quencher c 9 bp upstream of the fluorescent reporter dye, and a quencher (black hole quencher-1) on the 3’ end (Table 2). Optimal PCR conditions were evaluated in 15-µl reactions consisting of 7 µl of PCR supermix (termed IQSM), d 50 ng of ML09-121 gDNA, and nuclease-free water to volume. All possible combinations of 20, 10, and 5 pmol of each primer and 2, 1, and 0.5 pmol of probe were evaluated. Gradient PCR was performed to identify the highest annealing/extension temperature that still resulted in consistent amplification.
Primer and probe sequences used in the quantitative polymerase chain reaction (qPCR) specific for virulent Aeromonas hydrophila and the conventional PCR for the 16S ribosomal RNA (rRNA).

Tm = melting temperature; calculated as described previously. 11
Quantitative PCR
The VAh PCR was evaluated using 2 different commercial PCR supermixes, IQSM d for bacteria in broth culture and fish tissues and EMM e for environmental samples. For each supermix, the 15-μl PCR reactions contained 7 μl of PCR supermix, 20 pM of each primer, 2 pM of probe, and 5 μl of template DNA. Amplifications were performed on a real-time PCR platform f programmed for a 2-step amplification cycle based on the specific recommendations for each supermix. The reactions with the IQSM d supermix used a 3 min initial denaturation at 95°C followed by 40 cycles of 10 sec denaturation and 30 sec annealing/extension at 65°C. The reactions that used the EMM e supermix required a 15 min initial denaturation of 95°C followed by 40 cycles of 15 sec denaturation (95°C) and 1 min annealing/elongation at 65°C. Data collection was carried out following the annealing/extension step at the end of each cycle. The user-defined baseline relative fluorescence threshold was set at 10.0 for all analyses in this study.
PCR from intact cells
Nine different A. hydrophila (5 VAh; 4 RAh) isolates as well as isolates of Edwardsiella ictaluri (S97-773), Edwardsiella tarda (ATCC 15947), Escherichia coli (ATCC 25922), Enterobacter cloacae (ATCC 35030), Serratia odorifera (ATCC 33077), Aeromonas veronii (ML09-123), and Acinetobacter lwoffi (ATCC 17925) were streaked onto Mueller–Hinton agar a plates supplemented with 5% sheep blood and incubated for 24 hr at either 27°C (A. hydrophila, A. veronii, A. lwoffi, and E. cloacae) or 37°C (E. tarda, E. coli, and S. odorifera), with the exception of E. ictaluri, which was incubated at 27ºC for 48 hr. Individual colonies (n = 3) were picked from each isolate, resuspended in 100 µl of nuclease-free water, and then vortexed. A 5-µl aliquot of each suspension was used as template in individual reactions using EMM, e all of which were run on the same plate. On a separate plate, amplification with universal bacterial 16S ribosomal RNA (rRNA) gene-specific primers (Table 2) was performed on each sample to confirm the presence of bacterial DNA. The 15-µl reactions consisted of 7 µl of a commercial supermix, g 20 pmol of each primer, 5 µl of each colony suspension, and nuclease-free water to volume. The reaction parameters for the 16S rRNA gene analysis included an initial denaturation step of 95°C for 3 min followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/extension at 60°C for 2 min. Data collection was carried out following the annealing/extension step at the end of each cycle.
Sensitivity, repeatability, and reproducibility
To establish the sensitivity, linear dynamic range, repeatability, and reproducibility of the assay, 3 separate serial dilutions of ML09-121 gDNA (10–1 × 108 copies; estimated based on spectrophotometric quantification h and a genome size of 4,744,448 bp for A. hydrophila 12 ) were analyzed in triplicate on 3 separate occasions. In addition, 10-fold serial dilutions of known quantities of bacteria were quantified from 3 different VAh isolates. Briefly, cryostocks of 3 VAh isolates (AL09-74, AL09-79, and AL09-150) were streaked for isolation as described above. Individual colonies were grown in static BHI broth a at 27°C for 4 hr (early to mid-log phase), after which cultures were serially diluted 10-fold and plate counts were performed using the drop plate method. Aliquots from each dilution, ranging from 5 × 101 to 5 × 106 colony forming units (CFUs) were added to a 1.5-ml microcentrifuge tube and stored at −80°C until processing. Individual aliquots were analyzed in triplicate, which was repeated on 3 separate occasions. In addition to the gDNA isolated from each aliquot, each 96-well plate included a serial dilution of A. hydrophila (ML09-121) gDNA as a standard, ranging from 10 to 1 × 107 copies of target DNA (estimated as described previously) and 3 no-template controls.
Specificity and cross-reactivity
Overnight broth cultures for all A. hydrophila isolates (VAh and RAh) were pelleted by centrifugation at 16,000 × g for 3 min. Genomic DNA was isolated from bacterial cultures as previously described and quantified spectrophotometrically. Sample preparations were diluted to approximately 10 ng/µl using a commercial DNA hydration solution. i To test for variation in amplification from different VAh isolates, 50 ng of gDNA from each isolate was analyzed in triplicate on 3 separate occasions.
Construction of plasmid standard
A purified PCR amplicon was inserted into a plasmid vector to develop a stable standard that could be shared across different laboratories. Briefly, the PCR amplicon was generated in a 15-µl reaction consisting of 7 µl of IQSM, g 20 pmol of each primer, 0.5 pmol of probe, 50 ng of ML09-121 gDNA, and nuclease-free water to volume. Thermal cycling conditions consisted of an initial denaturation at 95°C for 3 min followed by 40 cycles of denaturation at 95°C for 15 sec and anneal/extension at 65°C for 1 min. The PCR amplicon was stained with ethidium bromide and visualized after electrophoretic migration under ultraviolet light. The product was gel excised by sharp dissection, purified, j then cloned and transfected into E. coli. k Plasmid DNA was purified, l linearized using SpeI restriction enzyme, m and quantified spectrophotometrically. For each PCR master mix, serial dilutions of the plasmid standard (7–7 × 105 copies) were analyzed in triplicate on 6 separate occasions to ensure consistency between mastermixes and stability of the plasmid.
Detection in broth culture and fish tissues
Individual cultures (n = 3) of VAh strain ML09-121 were grown without shaking at 27°C for 4 hr. Serial 10-fold dilutions were plated and counted in triplicate to approximate bacterial concentrations. Aliquots (50 µl) from each dilution were then added to a 1.5-ml microcentrifuge tube and stored at −80°C until further processed. Genomic DNA was isolated from aliquots (ranging from 101 to 105 CFU) using a commercial kit, b following the manufacturer suggested protocol for Gram-negative bacteria.
To determine the ability of the assay to detect VAh in fish tissues, individual cultures (n = 3) of VAh strain ML09-121 were grown and enumerated; aliquots were stored as described above. Posterior kidney biopsies (approximately 0.025 g) were collected from channel catfish reared for disease research at the holding facility of the National Warmwater Aquaculture Center (NWAC; Stoneville, Mississippi) and stored at −80°C until processing. Initially, catfish were confirmed PCR-negative for VAh by culture and the VAh qPCR. Aliquots (n = 3) from each individual broth culture dilution (ranging from 101 to 105 CFU) were added directly to individual kidney tissue samples prior to homogenization. Again, gDNA was isolated using a commercial kit, b following the manufacturer suggested protocol for animal tissues.
Genomic DNA of fish tissue samples, as well as straight broth culture aliquots, was used in the VAh PCR using IQSM d with the reaction parameters described above. For each plate, samples and no-template negative controls were analyzed in triplicate alongside concurrently run plasmid standards consisting of serial dilutions ranging from 7 to 7 × 105 copies, which were analyzed in duplicate.
Detection in environmental samples
Water (20 liters) and sediment (2 kg) collected from 3 commercial ponds with no previous history of the presence of the VAh strain were used to evaluate the sensitivity of the assay in environmental samples. Subsamples of sediment (approximately 0.25 g) were placed in 1.5-ml microcentrifuge tubes and stored at −80°C until analysis. The analysis of pond-water samples was performed according to previously published protocols for detection of pathogens in catfish pond water.3,4 Briefly, 40-ml subsamples were transferred to a 40-ml round-bottom centrifuge tube and centrifuged at 20,000 × g for 10 min. The supernatant was removed, and the pellet was resuspended in 1.5 ml of nuclease-free water and transferred to a 1.8-ml microcentrifuge tube. Aliquots (n = 3) ranging from 101 to 105 CFU of ML09-121 were added directly to individual sediment samples or pellets obtained from water samples. Total DNA was isolated from spiked environmental samples using a commercial kit n following the manufacturer suggested protocol for wet samples with the following modifications: 1) during the initial bead-beating step, samples were vortexed vigorously for 20 min; 2) no more than 500 µl of supernatant was transferred from the bead tube to the clean tube prior to the addition of 250 µl of solution C2. n Genomic DNA from environmental samples was analyzed using the EMM supermix e to account for potential inhibitors still present.
Assay variability
Water (20 liters) and sediment (2 kg) samples were collected from 5 different catfish ponds with no previous history of exposure to VAh. Environmental samples were processed as described previously. Posterior kidney biopsies (approximately 0.025 g) were collected from 5 channel catfish reared for disease research at NWAC and confirmed negative for VAh by culture and VAh qPCR. Aliquots (n = 5) of known quantities of ML09-121 (ranging from 2.4 × 105 to 2.4 × 106 CFU) were added directly to individual environmental samples or kidney samples. Genomic DNA for all environmental samples was analyzed using the EMM supermix, e while fish tissue samples were analyzed using the IQSM supermix. d
Results
Specificity and cross-reactivity of intact cells and gDNA in the VAh qPCR
Intact bacteria from colonies of the 5 VAh isolates tested demonstrated robust amplification, while no amplification was observed from colonies of the 4 RAh isolates, A. veronii, or any of the nonaeromonads (Table 3). Moreover, target DNA was consistently amplified from gDNA (50 ng) of all VAh isolates analyzed; however, amplification was not observed from gDNA of any RAh isolates (Table 4). With the exception of the 50 CFU aliquots, there was little variability (coefficient of variation [CV]: 1.6–11.1%) in amplification for similar quantities of bacteria from different VAh isolates (Table 5).
Results of the virulent Aeromonas hydrophila quantitative polymerase chain reaction (qPCR) analysis of intact cells.*
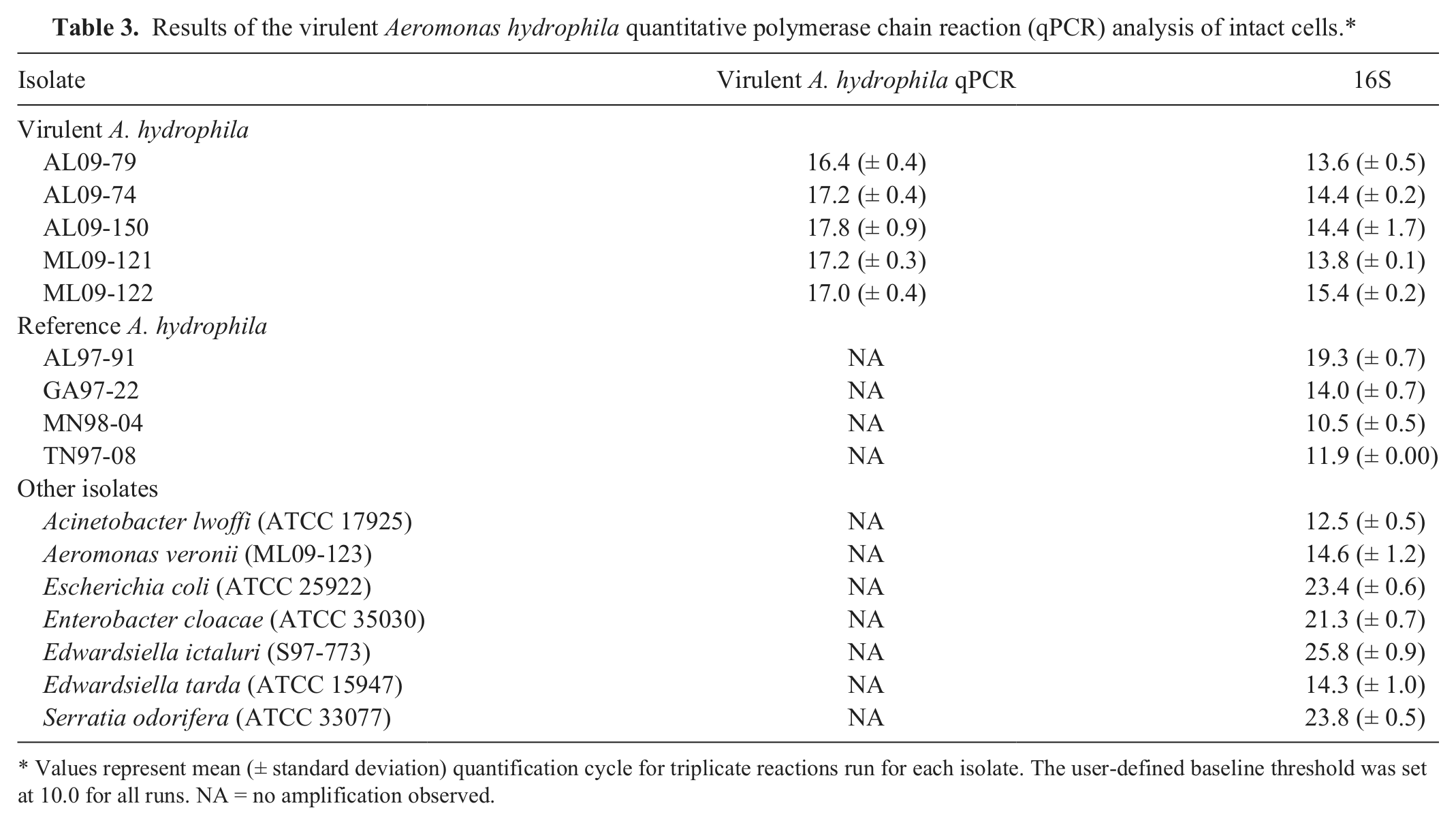
Values represent mean (± standard deviation) quantification cycle for triplicate reactions run for each isolate. The user-defined baseline threshold was set at 10.0 for all runs. NA = no amplification observed.
Results of the virulent Aeromonas hydrophila quantitative polymerase chain reaction analysis, demonstrating specificity of the primers against reference A. hydrophila isolates.*
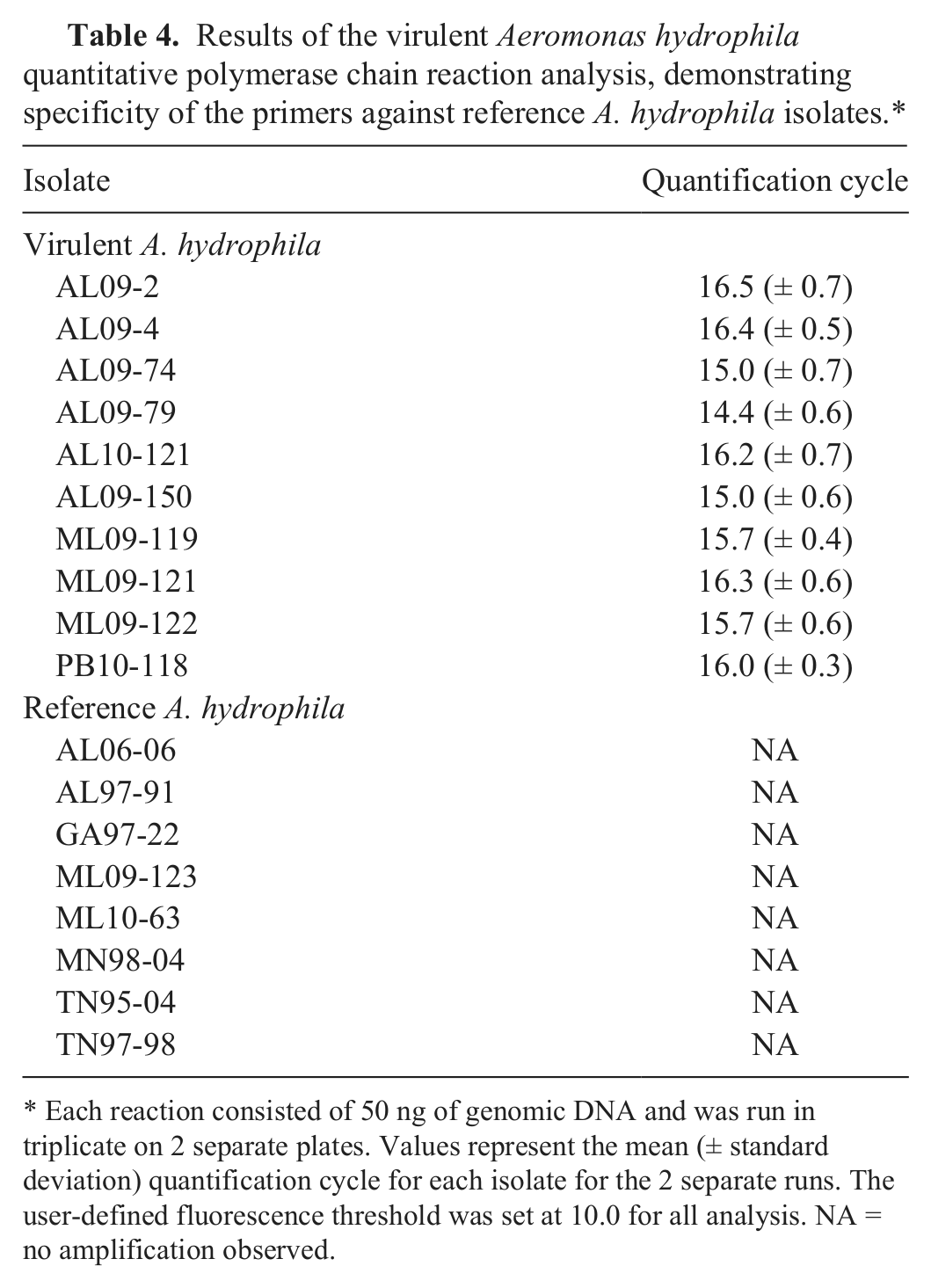
Each reaction consisted of 50 ng of genomic DNA and was run in triplicate on 2 separate plates. Values represent the mean (± standard deviation) quantification cycle for each isolate for the 2 separate runs. The user-defined fluorescence threshold was set at 10.0 for all analysis. NA = no amplification observed.
Amplification of target DNA from samples of known quantities of bacteria in the virulent Aeromonas hydrophila quantitative polymerase chain reaction.*
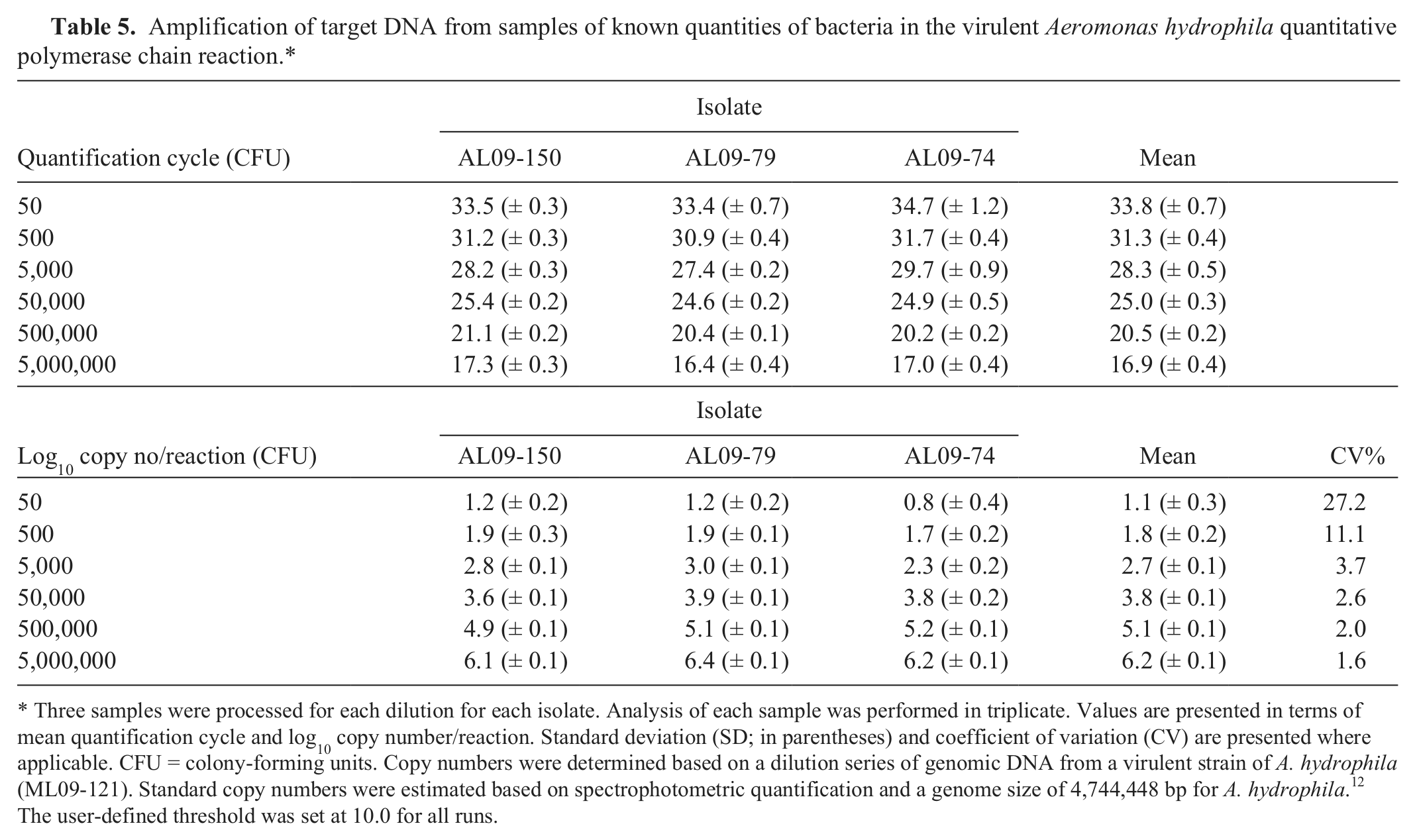
Three samples were processed for each dilution for each isolate. Analysis of each sample was performed in triplicate. Values are presented in terms of mean quantification cycle and log10 copy number/reaction. Standard deviation (SD; in parentheses) and coefficient of variation (CV) are presented where applicable. CFU = colony-forming units. Copy numbers were determined based on a dilution series of genomic DNA from a virulent strain of A. hydrophila (ML09-121). Standard copy numbers were estimated based on spectrophotometric quantification and a genome size of 4,744,448 bp for A. hydrophila. 12 The user-defined threshold was set at 10.0 for all runs.
Analytical sensitivity
Using a 10-fold serial dilution of gDNA, the assay was linear over 8 orders of magnitude and sensitive to an estimated 10 copies of target DNA (Fig. 1). Similar sensitivity was demonstrated using the linearized plasmid (approximately 7 copies of target DNA). Reactions with <7 copies of target DNA resulted in inconsistent amplification, often with no observed amplification in fewer than 40 cycles. For both qPCR supermixes, the assay was linear over 6 orders of magnitude ranging from 7 to 7 × 105 copies of the linearized plasmid standard, with similar curves observed between supermixes (Fig. 2).
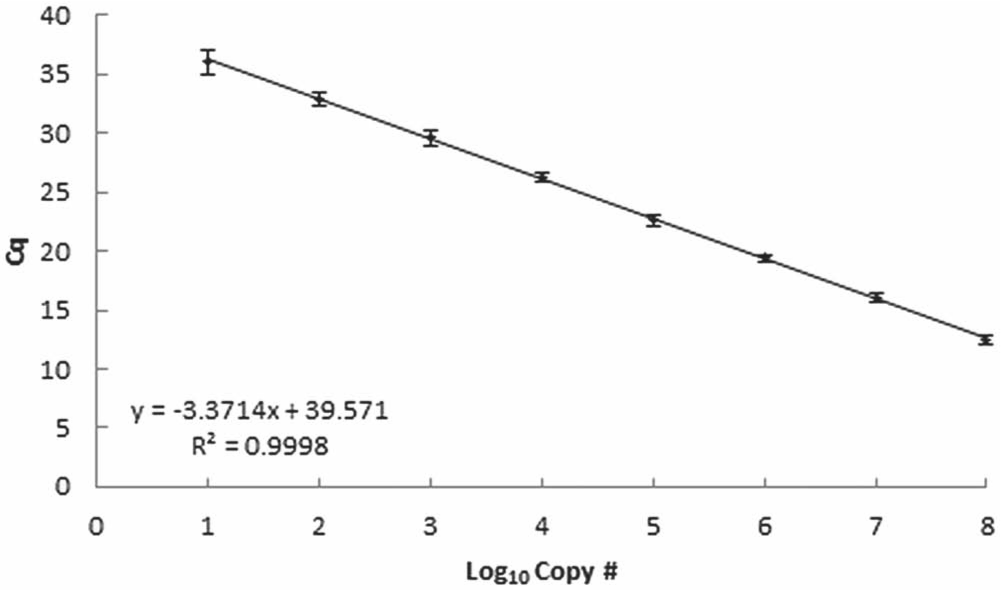
Results of virulent Aeromonas hydrophila quantitative polymerase chain reaction analysis of 3 separate 10-fold serial dilutions of genomic DNA isolated from 3 different cultures of a virulent strain of A. hydrophila (ML09-121). Values represent the mean (± standard deviation) quantification cycle (Cq) from triplicate reactions for each dilution run on 3 separate occasions. Approximate copy numbers per reaction were estimated based on spectrophotometric quantification and a genome size of 4,744,448 bp for A. hydrophila. 12 The user-defined threshold was set at 10.0 for all runs.
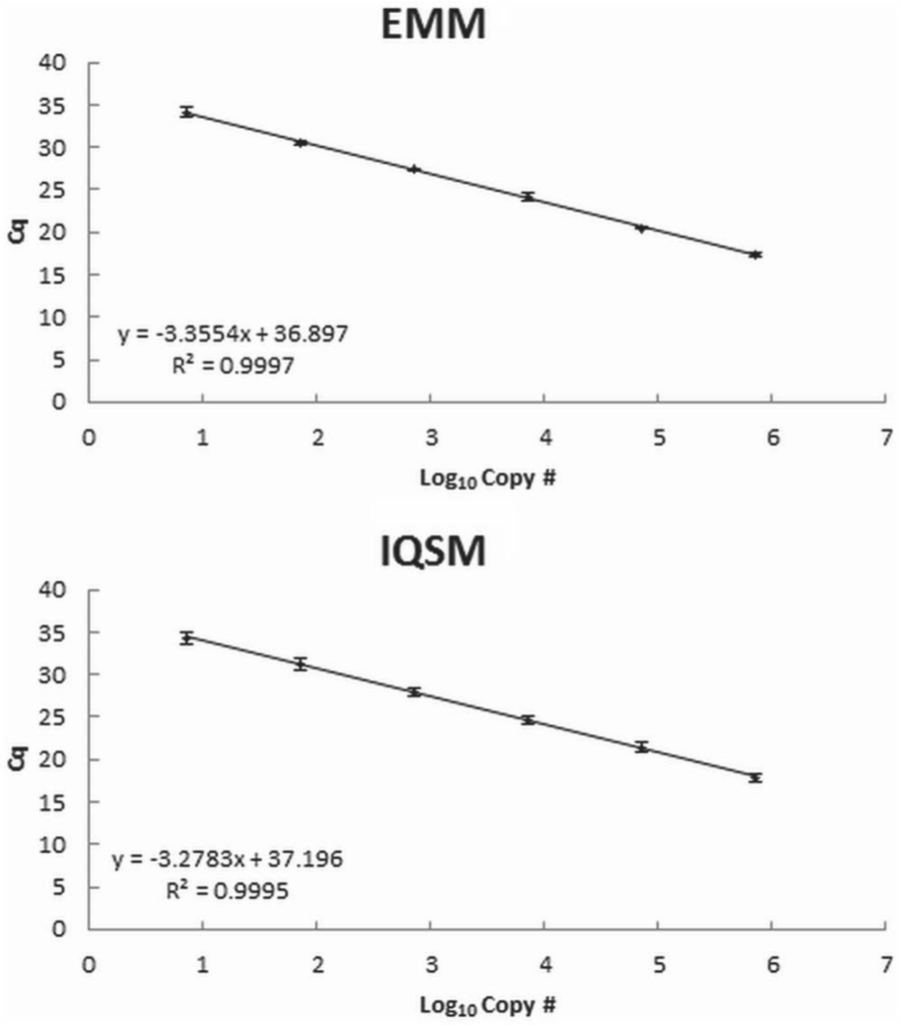
Comparison between polymerase chain reaction (PCR) supermixes,d,e EMM and IQSM, in a virulent Aeromonas hydrophila quantitative polymerase chain reaction using a 10-fold serial dilution of plasmid standard ranging from 7 to 7 × 105 copies of target DNA. Values represent the mean (± standard deviation) quantification cycle (Cq) for 6 different runs. The user-defined baseline threshold was set at 10.0 for all runs.
Clinical sensitivity and assay variability
The assay reliably detected target DNA in gDNA preparations from ~100 CFU/sample from broth culture. For samples with <100 CFU, amplification was inconsistent between replicates and occasionally absent, with 11% of reactions from aliquots of <100 CFU/sample giving negative results. When present, amplification resulted in quantification cycles (Cq) similar to those observed for approximately 100 CFU. The assay was linear over at least 5 orders of magnitude in these experiments (Fig. 3), indicating a clinical sensitivity of ~100 CFU/sample.
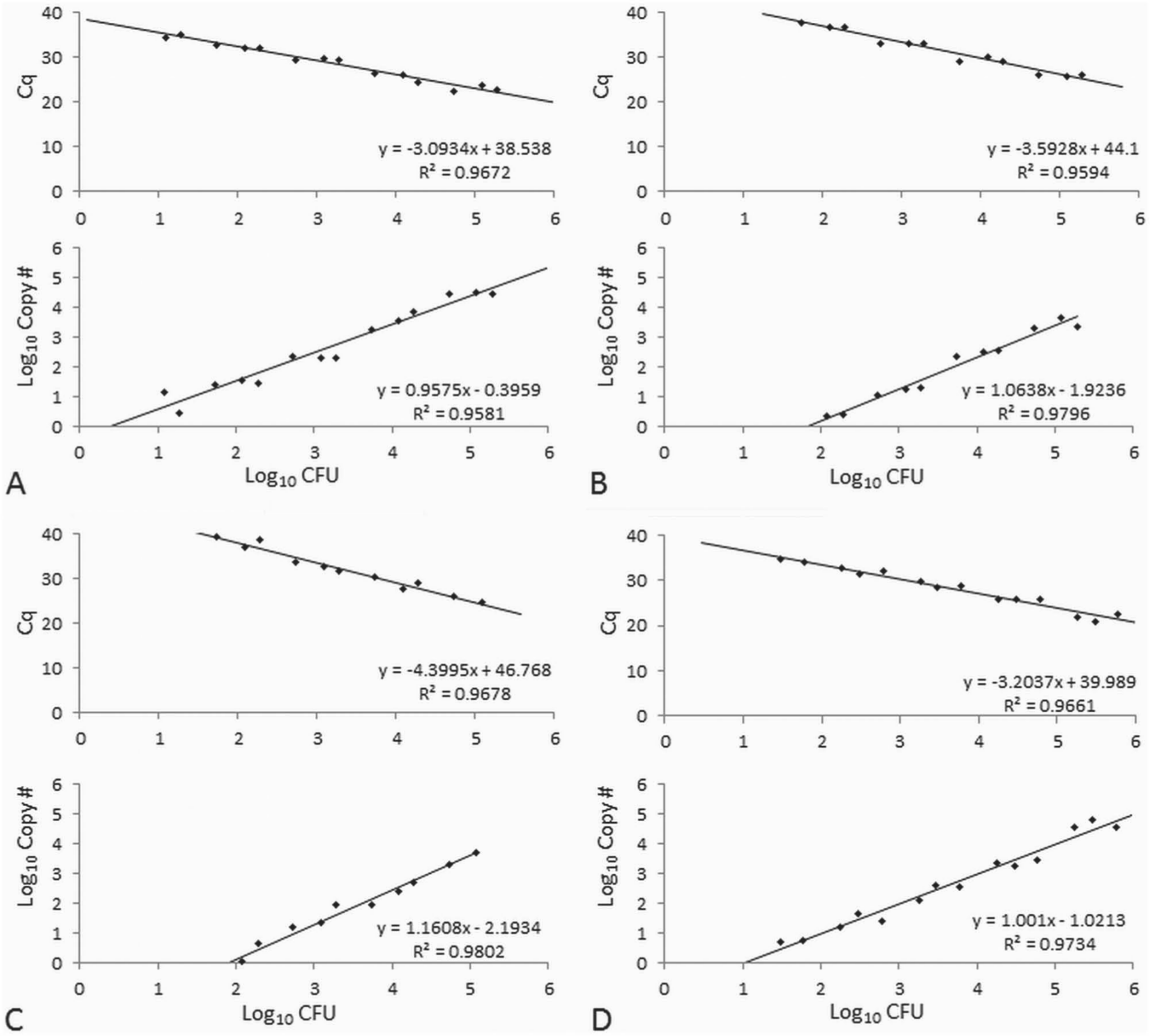
Results of virulent Aeromonas hydrophila quantitative polymerase chain reaction analysis of known quantities of a virulent strain of A. hydrophila grown in broth culture processed as is (
Similar to the broth culture results, the assay reliably detected (100% of reactions) and quantified approximately 100 CFU/sample from pond water and fish kidney tissues, demonstrating linearity over at least 5 orders of magnitude (Fig. 3). The assay was less sensitive in detecting bacteria out of sediment samples, with several reactions from aliquots of 100 CFU/sample yielding negative results. Reaction efficiencies (E) were estimated by the formula E = 10-1/s – 1, where s is the slope of the standard curve. Efficiencies ranged from 93% to 109% throughout the study, with a mean of 99.7%, well within the range of 90–110% (slope = 3.1–3.6) deemed acceptable in other assays.2,5,17 The assay was relatively consistent, with 2% CV in detecting similar quantities of bacteria from different kidney biopsies, with increased variability in pond water (5–18% CV) and sediment samples (7–16% CV; Table 6).
Results of virulent Aeromonas hydrophila quantitative polymerase chain reaction analysis of known quantities of bacteria added to kidney biopsies, pond water, or pond sediment samples.*
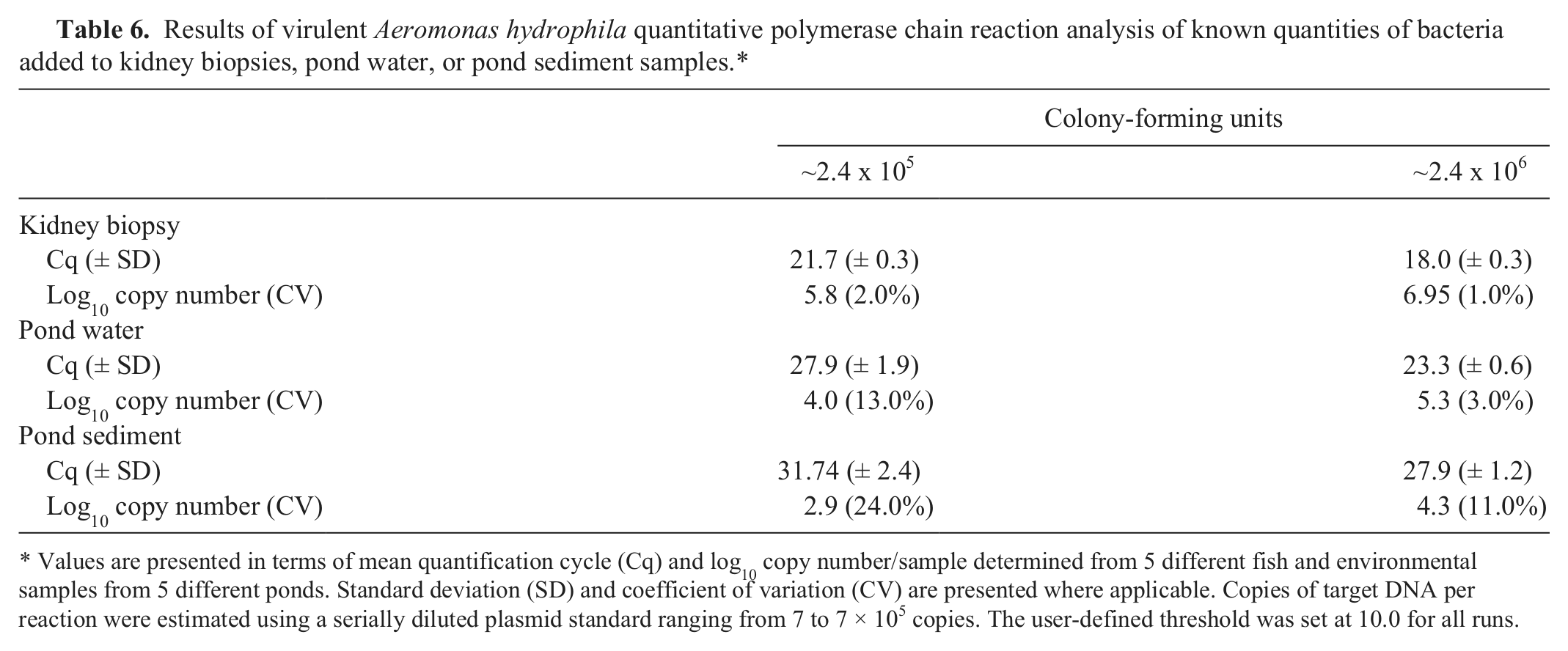
Values are presented in terms of mean quantification cycle (Cq) and log10 copy number/sample determined from 5 different fish and environmental samples from 5 different ponds. Standard deviation (SD) and coefficient of variation (CV) are presented where applicable. Copies of target DNA per reaction were estimated using a serially diluted plasmid standard ranging from 7 to 7 × 105 copies. The user-defined threshold was set at 10.0 for all runs.
Discussion
The protocols described herein provide an accurate and sensitive method for the rapid discrimination between the new VAh strains and the common strains of A. hydrophila that are routinely found in aquatic environments and as normal flora of freshwater fish. There was consistent amplification of all isolates identified as the highly virulent strain, while there was no amplification from the more common A. hydrophila isolates or other Gram-negative bacteria, including the ubiquitous catfish pathogens E. ictaluri and E. tarda.
Triplicate serial dilutions of gDNA analyzed on separate plates demonstrated that the assay was reproducible and that the linear dynamic range covered at least 8 orders of magnitude with a sensitivity of ≥7 copies of target DNA. Genomic DNA isolated from similar quantities of bacteria demonstrated low variability between replicate preparations from multiple isolates, demonstrating a level of precision within the quantifiable range of the assay (CV was <15% for >100 CFU) that has been considered acceptable in similar assays. 3
Once suspect colonies are identified on a culture plate, positive identification can be obtained in less than 2 hr, providing a confirmatory diagnosis in as little as 24 hr from initial submission. Furthermore, the assay was proven to be reliable in detection and quantification of VAh in channel catfish kidney tissues. Given the rapid onset of mortality associated with VAh, a rapid diagnostic test will shorten response time in cases where aggressive treatments and increased biosecurity are required.
Quantitative PCR has been used to detect a vast array of pathogens in aquatic environments,1,6,14 but amplification of targets from catfish ponds is complicated by the presence of humic acids and other compounds inhibitory to the reaction.3,4,16 Moreover, efficiency of DNA isolation is subject to a high degree of variability based on the types and amounts of organic materials present. Previous work has demonstrated that reducing the volume of water samples can reduce variability between water samples from different ponds.
3
Similarly, when analyzing sediments, larger samples (>0.25 g) may result in less efficient DNA isolation. Clay particulates can make samples unwieldy, making physical manipulation of the sample more difficult, resulting in greater variability between samples. Conversely, sediment samples that consist of wetter, finer sediment or water samples that contain few suspended solids are easier to manipulate, as water makes up a larger component of their mass. Regardless, using 40 ml of pond water or approximately 0.25 g of sediment, the assay in the current study was able to consistently detect and estimate the quantity of VAh from a variety of substrates from different ponds, with a sensitivity limit of
At present, the prevalence of this strain of A. hydrophila in commercial culture systems is not known, along with the temporal dynamics of bacterial populations in aquaculture pond water and sediments in the days or weeks leading up to an outbreak. In addition to the diagnostic benefits of this assay, detection and quantification of VAh from pond water, sediment, and fish tissues are valuable tools for tracking bacteria between ponds and operations, providing finer resolution for epidemiological studies to help identify factors that predispose fish populations to outbreaks. Likewise, the ability to quantify the bacteria in the environment also provides means to measure the efficacy of vaccination, antibiotic therapies, chemotherapeutants, and other treatment regimens toward reducing bacterial populations and minimizing losses. This newly developed qPCR assay could be a critical tool for researchers and diagnosticians aiming to reduce losses attributable to this emerging, highly pathogenic strain of A. hydrophila.
Footnotes
Acknowledgements
This study is Mississippi Agricultural and Forestry Experiment Station (MAFES) publication no. J-12271.
a.
BD Diagnostic Systems, Sparks, MD.
b.
DNeasy Blood and Tissue Kit, Qiagen GmBH, Hilden, Germany.
c.
ZEN, Integrated DNA Technologies, Coralville, IA.
d.
iQ Supermix, Bio-Rad Laboratories, Hercules, CA.
e.
TaqMan Environmental Mastermix v. 2.0, Applied Biosystems, Carlsbad, CA.
f.
iCycler CFX-96 Touch, Bio-Rad Laboratories, Hercules, CA.
g.
SsoFast EvaGreen Supermix, Bio-Rad Laboratories, Hercules, CA.
h.
NanoDrop 2000, NanoDrop Technologies, Wilmington, DE.
i.
Puregene DNA Hydration Solution, Qiagen GmBH, Hilden, Germany.
j.
QIAquick Gel Extraction kit, Qiagen GmBH, Hilden, Germany.
k.
Topo-TA cloning kit for sequencing, Invitrogen Corp., Carlsbad, CA.
l.
Qiaprep Spin Miniprep Kit, Qiagen GmBH, Hilden Germany.
m.
Promega Corp., Madison, WI.
n.
Powersoil DNA Isolation Kit, Mo Bio Laboratories Inc., Carlsbad, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research was supported by the Mississippi State University College of Veterinary Medicine, the Mississippi Agricultural and Forestry Experiment Station, the Alabama Agricultural Experiment Station, Auburn University (Hatch project no. ALA021-1-09005), and the USDA-ARS Catfish Health Initiative (CRIS 6402-31320-002-02).