
Abstract
Pen-based oral fluid sampling has proven to be an efficient method for surveillance of infectious diseases in swine populations. To better interpret diagnostic results, the performance of oral fluid assays (antibody- and nucleic acid-based) must be established for pen-based oral fluid samples. Therefore, the objective of the current study was to determine the probability of detecting Porcine reproductive and respiratory syndrome virus (PRRSV) infection in pen-based oral fluid samples from pens of known PRRSV prevalence. In 1 commercial swine barn, 25 pens were assigned to 1 of 5 levels of PRRSV prevalence (0%, 4%, 12%, 20%, or 36%) by placing a fixed number (0, 1, 3, 5, or 9) of PRRSV-positive pigs (14 days post PRRSV modified live virus vaccination) in each pen. Prior to placement of the vaccinated pigs, 1 oral fluid sample was collected from each pen. Thereafter, 5 oral fluid samples were collected from each pen, for a total of 150 samples. To confirm individual pig PRRSV status, serum samples from the PRRSV-negative pigs (n = 535) and the PRRSV vaccinated pigs (n = 90) were tested for PRRSV antibodies and PRRSV RNA. The 150 pen-based oral fluid samples were assayed for PRRSV antibody and PRRSV RNA at 6 laboratories. Among the 100 samples from pens containing ≥1 positive pig (≥4% prevalence) and tested at the 6 laboratories, the mean positivity was 62% for PRRSV RNA and 61% for PRRSV antibody. These results support the use of pen-based oral fluid sampling for PRRSV surveillance in commercial pig populations.
Surveillance for infectious diseases in swine populations is a key component in the prevention and/or control of clinical losses, but is often limited by the cost and inconvenience of collecting individual pig samples. Under both experimental and field conditions, oral fluid samples have proven to be effective for the detection of a variety of pathogens (e.g., Influenza A virus, Porcine circovirus-2, and Porcine reproductive and respiratory virus [PRRSV]).6,7,9-12 Oral fluid sampling is possible because it is well matched with inherent pig behavior. Exploration is part of porcine normal behavior, and pigs instinctively test new objects by chewing. 8 Animal behaviorists report that pigs prefer flexible, destructible, chewable objects 2 (i.e., adjectives that aptly describe rope; Zonderland J, Vermeer M, Ter Avest A, et al.: 2001, Measuring a pig’s preference for suspended toys by using an automated recording technique. In: Proceedings of the International Symposium of the C.I.G.R. Animal Welfare Considerations in Livestock Housing Systems, 2nd Technical Section, pp. 147–156. Szlarska Poreba, Poland, October 23–25). These behavioral imperatives operate even during acute infection, hence sample collection success rates remain unchanged during acute infection with PRRSV 5 and Influenza A virus (Millman S, Brooks R Jr, Zimmerman J, et al.: 2009, Role of behavior in non-invasive disease surveillance of swine influenza virus. J Anim Sci 87:E-Suppl:ii).
Options for PRRSV surveillance using oral fluid specimens include real-time reverse transcription polymerase chain reaction (real-time RT-PCR) for the detection of nucleic acids and antibody detection assays.5-7,9-12 While PCR-based assays are useful for detecting the immediate circulation of pathogens, antibody-based assays are informative regarding herd immunity and the history of an infection in the population. Because both assays reflect different phases of infection, the best test can be selected to serve the need. Although both approaches are considered diagnostically sensitive and specific, to the authors’ knowledge, test performance using oral fluid samples collected at the pen level has not been reported. Therefore, the objective of the current study was to determine the probability of detecting PRRSV or anti-PRRSV antibody in pen-based oral fluid samples from pens of known PRRSV prevalence.
Materials and methods
Experimental design
In 1 commercial swine barn, 25 pens holding 25 pigs each were randomly assigned a to 1 of 5 levels of PRRSV prevalence (0%, 4%, 12%, 20%, or 36%). Pen-based oral fluid samples collected from each pen and serum collected from each pig were assayed for PRRSV antibody and PRRSV RNA. Thereafter, the probability of detecting a PRRSV-positive pen by antibody enzyme-linked immunosorbent assay (ELISA) or real-time RT-PCR using 1 pen-based oral fluid sample was modeled as a function of within-pen PRRSV prevalence using logistic regression. b The current study was approved by the Iowa State University Office for Responsible Research (#8-11-7201-S).
Logistics and study timeline
As shown in Table 1, PRRSV-negative nursery age pigs (n = 90) located in Missouri were intramuscularly vaccinated with 2.0 ml of a PRRSV modified live vaccine c on December 23. On January 2, the vaccinated pigs were transported to a commercial finishing site in Iowa and placed in isolation.
Project timeline describing the logistics and experimental design of the current study detecting Porcine reproductive and respiratory syndrome virus (PRRSV) infection using pen-based swine oral fluid specimens as a function of within-pen prevalence.

Ingelvac PRRS MLV, Boehringer Ingelheim Vetmedica Inc., St. Joseph, MO.
On January 3, a separate group of PRRSV-negative nursery age pigs (n = 535) from Oklahoma was transported to the Iowa site and placed in the study barn. On January 4, these pigs were sorted into the 25 study pens, after which 1 oral fluid sample was collected from each pen (negative control samples). On the morning of January 5, serum samples were collected from the 535 Oklahoma pigs in the 25 pens for the purpose of authenticating their individual PRRSV status.
On the afternoon of January 5, the 90 PRRSV-vaccinated Missouri pigs were moved to the study barn and placed in each pen. The number of pigs sorted into each pen was determined by the PRRSV prevalence randomly assigned a to the pen (i.e., 0%, 4%, 12%, 20%, or 36%). That is, PRRSV prevalence was established by placing a fixed number (0, 1, 3, 5, or 9) of viremic and/or antibody-positive pigs in pens such that the combination of negatives and positives in each pen totaled 25 pigs.
On the morning of January 6 (i.e., 14 days postvaccination), 5 successive pen-based oral fluid samples (30-min sampling) were collected from each pen. After the fifth oral fluid sampling, serum samples were collected from each of the 90 PRRSV-vaccinated c pigs to establish the PRRSV status of all vaccinated pigs.
Biological samples
Serum specimens were collected using a single-use blood collection system d and serum separation tubes. d Blood samples were allowed to clot at room temperature and centrifuged for 10 min at 1,000 × g to separate the serum. All samples were stored in 2-ml cryogenic vials at −70°C until shipped for testing.
Pen-based oral fluid specimens were collected as previously described.5,10,11 Briefly, approximately 100 cm of 3-strand (1.6 cm) cotton rope e was suspended in the pen for 30 min to allow the pigs to interact with the rope. After 30 min, the saturated (wet) portion of the rope was placed in a plastic bag, severed from the dry portion of the rope, and sealed in the plastic bag. To harvest the oral fluid sample, each bag with the rope inside was pressed through a hand wringer. f Oral fluid pooled in the corner of the bag, the bag was cut, and oral fluid was decanted into 50-ml centrifuge tubes. Samples were aliquoted and stored in 2-ml cryogenic vials at −70°C until shipped for testing.
Diagnostic assays
All serum and oral fluid sample sets were randomly ordered a and tested for PRRSV antibodies g and PRRSV RNA at a total of 8 participating laboratories. Oral fluid (n = 150) and vaccinated pig serum (n = 90) sample sets were tested for PRRSV antibodies at laboratories 1–3 and 6–8, and PRRSV RNA at laboratories 1–6. Serum samples from expected PRRSV-negative Oklahoma pigs (n = 535) were tested individually for PRRSV antibody at laboratories 1 and 8.
For PRRSV real-time RT-PCR testing, the expected PRRSV-negative Oklahoma pig serum samples (n = 535) were pooled by fives at the time of processing, thereby creating 107 pooled samples. Pooled samples were tested for PRRSV RNA at laboratories 4 and 5. If a pooled sample tested positive, the samples that composed the respective pool were tested individually by the laboratory.
PRRSV antibody ELISA
The 6 laboratories (1–3, 6–8) testing serum and oral fluid samples for PRRSV antibodies used the same commercial ELISA assay g and followed the same testing protocols. Serum was tested according to the manufacturer’s instructions. Oral fluid specimens were tested using a PRRSV immunoglobulin G (IgG) antibody ELISA based on a commercial ELISA.g,4,6,7 In brief, the PRRSV oral fluid IgG antibody ELISA was performed by diluting oral fluid samples 1:2 in sample diluent provided in the kit, then transferring 250 μl of diluted sample onto the kit antigen plates. Negative and positive kit controls were diluted 1:30 using sample diluent, and 100 μl was added to the plates in duplicate. Plates were incubated at 4°C for 16 hr (overnight) and then washed 3 times using 400 μl of 1× wash solution per well. Horseradish peroxidase–conjugated anti-pig IgGFC h antibody (100 μl) was added and the plates incubated at 22°C for 30 min. After washing the plates 3 times, 100 μl of tetramethylbenzidine was added to all wells and the plates incubated for 15 min at 22°C. The color-developing reaction was stopped by pipetting 100 μl of kit stop solution into each well, after which plates were read at 650 nm. Results were reported as sample-to- positive ratios (S/P), with responses ≥0.40 considered positive.
PRRSV RT-PCR
Six laboratories (1–6) tested oral fluid (n = 150) and vaccinated pig serum (n = 90) sample sets. Two laboratories (4 and 5) tested the pooled samples from the expected PRRSV-negative Oklahoma pig serum samples (n = 107). Laboratory-specific PRRSV real-time RT-PCR assay protocols are described in the following sections.
Laboratory 1
Total RNA was extracted from serum and oral fluid using a commercial extraction kit. i Serum was extracted following the manufacturer’s instructions, and oral fluid was extracted using a high volume modified lysis protocol (protocol A2). 1 The lysis/binding solution for protocol A2 was prepared by adding 200 μl of carrier RNA to 45 ml of lysis/binding solution without the addition of isopropanol. For the lysis step, 300 μl of sample was added to 450 μl of lysis/binding solution, vortexed for 3 min, and centrifuged at 2,500 × g for 6 min. A volume of 600 μl of lysate was added to a mixture of 350 μl of isopropanol and 20 μl of magnetic bead mix prior to extraction and then eluted into 90 μl of buffer. The protocol used 300 μl and 450 μl of wash solutions I and II, respectively. The extraction was completed with an automated magnetic particle processor j using programs AM_1836_DW_50_v3 and AM1836_DW_HV_v3 for serum and oral fluid, respectively. Two positive controls and 1 negative control were included with each extraction.
The PRRSV real-time RT-PCR was performed with a commercial reagent set. k An internal control l was included in the master mix to monitor RT-PCR amplification and to detect failed RT-PCR reactions. All extraction controls and 1 negative amplification control were included with each RT-PCR run. Master mix component volumes per well for serum testing consisted of 12.5 μl of 2× RT-PCR buffer, 2.5 μl of 10× PRRSV primer probe mix, 1.25 μl of 20× multiplex RT-PCR enzyme mix, 0.35 μl of 100 copies/μl internal control, l and 0.4 μl of nuclease-free water. For oral fluid testing, reagent volumes were the same as serum, with the exception of 2.5 μl of 20× multiplex RT-PCR enzyme mix and 0.5 μl of nuclease-free water. Ultimately, 17 μl of master mix was combined with 8 μl of serum RNA extract or 18 μl of master mix was combined with 7 μl of oral fluid extract onto a 96-well plate. The reaction was completed using the following thermal cycling conditions: 1 cycle at 45°C for 10 min, 1 cycle at 95°C for 10 min, and 40 cycles of 97°C for 2 sec and 60°C for 40 sec.
Laboratory 2
Total RNA was extracted from serum and oral fluid using a commercial extraction kit. i Serum was extracted following the manufacturer’s instructions, and oral fluid was extracted using a high volume modified lysis protocol (protocol A2). 1 The lysis/binding solution for protocol A2 was prepared by adding 623 μl of carrier RNA to 40 ml of lysis/binding solution without the addition of isopropanol. For the lysis step, 300 μl of sample was added to 450 μl of lysis/binding solution, vortexed for 3 min, and centrifuged at 16,000 × g for 2 min. A volume of 600 μl of lysate was added to a mixture of 350 μl of isopropanol and 20 μl of magnetic bead mix prior to extraction and then eluted into 90 μl of buffer. The extraction protocol used 300 μl and 450 μl of wash solutions I and II, respectively. The extraction was completed with an automated magnetic particle processor j using program AM1836_DW_HV_v3.
The PRRSV real-time RT-PCR was performed with a commercial reagent set, m as directed by the manufacturer. The reaction was completed using the following thermal cycling conditions: 1 cycle at 48°C for 15 min, 1 cycle at 95°C for 2 min, and 45 cycles of 95°C for 5 sec and 60°C for 40 sec.
Laboratory 3
Total RNA was extracted using commercial extraction kits for serum i and oral fluid. n Serum was extracted following the manufacturer’s instructions and oral fluid was extracted following the protocol for “all other sample types.” The extraction was completed using an automated particle processor. o
The PRRSV real-time RT-PCR was performed with a commercial reagent set. p Master mix component volumes per well for serum and oral fluid testing consisted of 12.5 μl of 2× RT-PCR buffer, 2.5 μl of 10× PRRSV primer probe mix, 2.5 μl of 20× multiplex RT-PCR enzyme mix, and 0.35 μl of 100 copies/μl of internal control. l Ultimately, 18 μl of master mix (rounded up) was combined with 7 μl of RNA extract onto a 96-well PCR plate. The reaction was completed using the following thermal cycling conditions: 1 cycle at 45°C for 10 min, 1 cycle at 95°C for 10 min, and 40 cycles of 95°C for 2 sec and 60°C for 45 sec.
Laboratory 4
Total RNA was extracted from serum and oral fluid using a commercial extraction kit. i For serum samples, lysis/binding solution was prepared by combining the lysis/binding solution concentrate from the kit with an additional volume of lysis/binding solution, q without the addition of isopropanol. Two microliters of carrier RNA was then added to 350 μl of the prepared lysis/binding solution concentrate and 350 μl of 100% isopropanol per reaction. Wash solution I was prepared by adding 12 ml of 100% isopropanol to the wash solution I concentrate, and adding 70 ml of 100% isopropanol to an additional volume of wash solution I concentrate. r Both were then combined to create the final wash solution I. Wash solution II was prepared by adding 32 ml of 100% ethanol to the wash solution II concentrate, and adding 160 ml of 100% ethanol to an additional volume of wash solution II concentrate. s Both were then combined to create the final wash solution 2. Three hundred microliters of sample was added to 700 μl of prepared lysis/binding solution prior to extraction and then eluted into 90 μl of buffer. The protocol used 300 μl and 450 μl of wash solutions I and II, respectively. The extraction was completed with an automated particle processor o using program AM1836_DW _HV_v3.
For oral fluid samples, lysis/binding solution was prepared by adding 2 μl of carrier RNA and 2 μl of internal control l to 450 μl of lysis/binding solution per reaction, without the addition of isopropanol. For the lysis step, 300 μl of sample was added to 450 μl lysis/binding solution, vortexed for 5 min, and centrifuged at 3,300 × g for 5 min. A volume of 600 μl of lysate was added to 350 μl of isopropanol and 20 μl of bead mix prior to extraction and then eluted into 90 μl of buffer. The protocol used 300 μl and 450 μl of wash solutions I and II, respectively. The extraction was completed with an automated magnetic particle processor p using program AM1836_DW_300v2.
The PRRSV real-time RT-PCR was performed with a commercial reagent set. t Master mix component volumes per well for serum and oral fluid testing consisted of 12.5 μl of 2× RT-PCR buffer, 2.5 μl of 10× PRRSV primer probe mix, 2.5 μl of 10× multiplex RT-PCR enzyme mix, and 0.5 μl of nuclease-free water. Ultimately, 18 μl of master mix was combined with 7 μl of RNA extract onto a 96-well plate. The reaction was completed using the following thermal cycling conditions: 1 cycle at 48°C for 10 min, 1 cycle at 95°C for 10 min, and 40 cycles of 95°C for 15 sec and 60°C for 70 sec.
Laboratory 5
Total RNA was extracted from serum and oral fluid samples using a commercial extraction kit. n Serum samples were extracted following the manufacturer’s instructions for low cell content samples. Oral fluid samples were extracted using the protocol for oral fluid samples. Three hundred microliters of sample was used per extraction, and reagents were prepared according to the manufacturer’s instructions with 1 exception: in place of 2 μl of internal control l in the lysis/binding solution, 8 μl of in-house extraction/inhibition control t was used. Extraction of both sample types was completed with an automated particle processor u using program 4462359_DW_HV.
The PRRSV real-time RT-PCR was performed with a commercial reagent set. m Serum component volumes per well included 19.25 μl of master mix (includes buffer, primer, and probes), 0.25 μl of enzyme 1, 0.50 μl of enzyme 2, and 5 μl of extracted serum sample. Oral fluid component volumes per well included 16.25 μl of master mix (includes buffer, primer, and probes), 0.25 μl of enzyme 1, 0.50 μl of enzyme 2, and 8 μl of extracted oral fluid sample. Plates were centrifuged before being loaded onto the thermocycling instrument. The reaction was completed using the following thermal cycling conditions: 1 cycle at 48°C for 15 min, 1 cycle at 95°C for 2 min, and 45 cycles of 95°C for 5 sec and 60°C for 40 sec.
Laboratory 6
Total RNA was extracted from serum and oral fluid using a commercial extraction kit. i Serum was extracted following the manufacturer’s instructions with the following exceptions: a volume of 100 μl of sample was used with 300 μl of wash solution I, 450 μl of wash solution II, and 75 μl of elution buffer. Oral fluid was extracted using a modified protocol. Lysis/binding solution was prepared by adding 1,278 μl of carrier RNA to 100 ml of lysis/binding solution concentrate. q For the lysis step, 300 μl of sample was added to 400 μl of lysis/binding solution and processed using protocol AM1836_DW_saliva with an automated magnetic particle processor. o Plates were then centrifuged for 6 min at 6,000 × g. A volume of 600 μl of supernatant from the centrifuged sample/lysis solution was then added to a mixture of 350 μl of isopropanol and 20 μl of bead mix prior to elution into 75 μl of elution buffer. The protocol used 300 μl and 450 μl of wash solution I and II, respectively. The extraction was completed with an automated magnetic particle processor o using program AM1836_100_custom.
The PRRSV real-time RT-PCR was performed with a commercial reagent set. k Master mix component volumes per well for serum and oral fluid testing consisted of 12.5 μl of 2× RT-PCR buffer, 2.5 μl of 10× PRRSV primer probe mix, 2.5 μl of 20× multiplex RT-PCR enzyme mix, and 0.5 μl of internal control. l Ultimately, 18 μl of master mix was combined with 7 μl of RNA extract onto a 96-well plate and centrifuged at 2,000 × g for 5 sec. The reaction was completed using the following thermal cycling conditions: 1 cycle at 48°C for 10 min, 1 cycle at 95°C for 10 min, and 40 cycles of 97°C for 2 sec and 60°C for 40 sec.
Statistical analysis
Analyses were performed to compare laboratory performance and model the probability of a PRRSV-positive oral fluid sample as a function of within-pen prevalence. Interlaboratory comparisons were performed on qualitative (Cochran Q) v and quantitative (analysis of variance [ANOVA]) b results, and the effect of sampling order was compared on qualitative data using Chochran Q v test. The ANOVA model was a mixed model using both pen and pig as random effects. All laboratories used the same PRRSV antibody ELISA protocol and cutoff (S/P value ≥ 0.40). The ELISA S/P values were log-transformed to meet the criterion of normal distribution prior to quantitative analyses. The PRRSV real-time RT-PCR protocols varied among laboratories, as did threshold cycle cutoffs. Qualitative analyses were based on cutoffs established by each laboratory. The probability of a RT-PCR– or ELISA-positive oral fluid test was modeled using a repeated measures logistic regression b model with pen as a random effect. In the logistic model, qualitative assay results were modeled as the response variable and the actual within-pen PRRSV prevalence as the explanatory variable.
Results
The PRRSV ELISA testing of serum samples (n = 535) from the expected PRRSV-negative Oklahoma pigs produced 1 positive specimen at each of the 2 laboratories testing these samples. This sample was ELISA positive on retest. All ELISA results from this pen were omitted from the ELISA analyses because the response in the unexpected PRRSV-positive pig could not be accounted for. The PRRSV real-time RT-PCR of pooled serum (n = 107) from these animals was negative at the 2 laboratories performing the assays.
Qualitative and quantitative PRRSV real-time RT-PCR and ELISA responses for vaccinated pigs (n = 90) are listed in Table 2 for individual laboratories. Analyses of vaccinated pig serum testing results showed statistically significant (P ≤ 0.05) variation in qualitative PRRSV real-time RT-PCR results among laboratories, but not qualitative ELISA results. Quantitative results from both assays showed significant differences (P ≤ 0.05) among laboratories. Three vaccinated pigs were PRRSV antibody ELISA negative at the 6 laboratories testing these samples; 1 of these 3 pigs also tested PRRSV real-time RT-PCR negative at 6 laboratories. The unanimous testing results were used to establish the actual within-pen PRRSV prevalence for subsequent data analyses.
Results of testing serum samples collected from 90 pigs 14 days after vaccination with a Porcine reproductive and respiratory syndrome virus (PRRSV) modified live vaccine.*
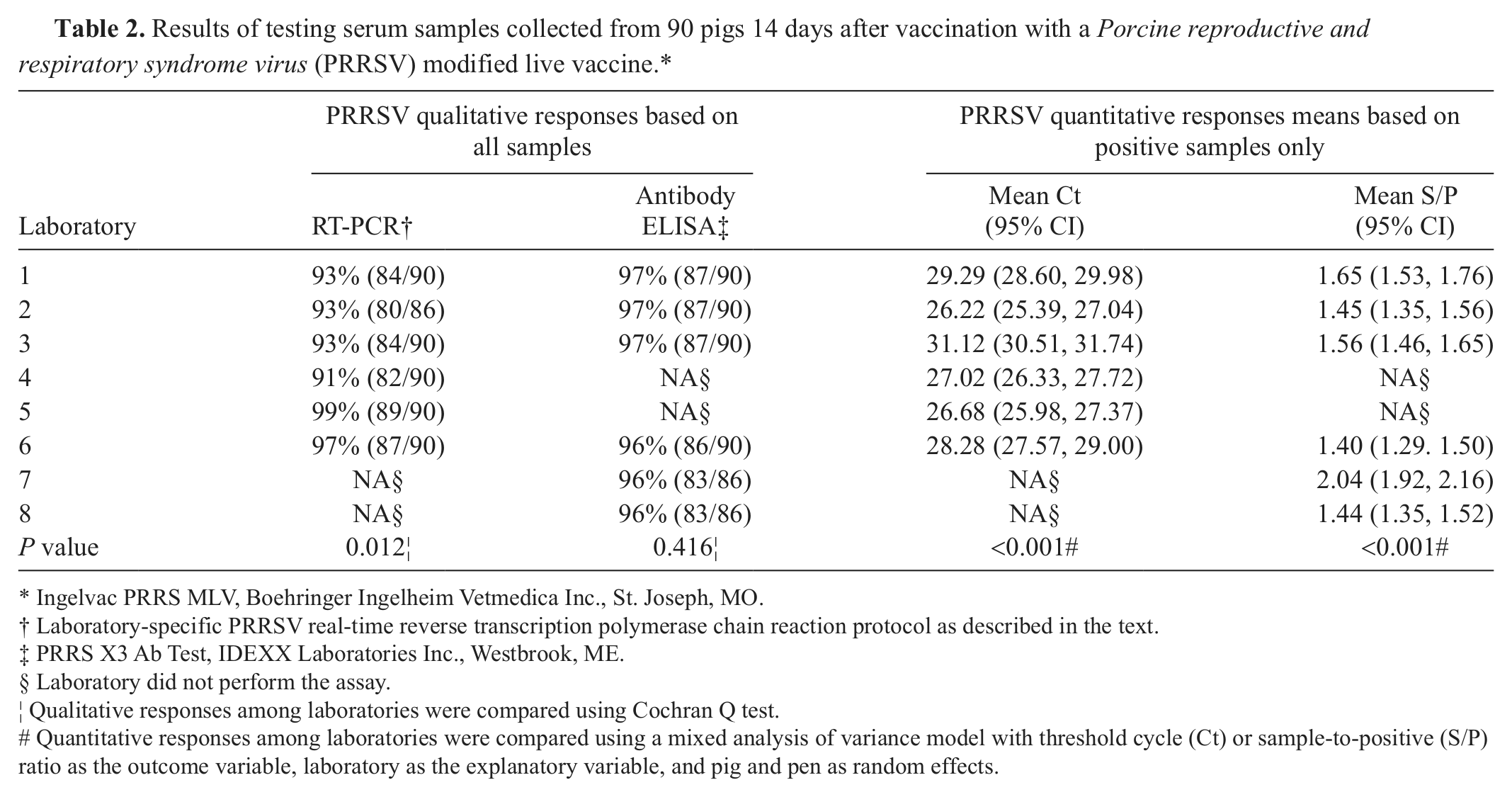
Ingelvac PRRS MLV, Boehringer Ingelheim Vetmedica Inc., St. Joseph, MO.
Laboratory-specific PRRSV real-time reverse transcription polymerase chain reaction protocol as described in the text.
PRRS X3 Ab Test, IDEXX Laboratories Inc., Westbrook, ME.
Laboratory did not perform the assay.
Qualitative responses among laboratories were compared using Cochran Q test.
Quantitative responses among laboratories were compared using a mixed analysis of variance model with threshold cycle (Ct) or sample-to-positive (S/P) ratio as the outcome variable, laboratory as the explanatory variable, and pig and pen as random effects.
Qualitative and quantitative PRRSV real-time RT-PCR and ELISA testing results for the oral fluid samples from pens of known prevalence are reported in Tables 3 and 4. Analyses of qualitative oral fluid testing results showed statistically significant differences (P ≤ 0.05) in detection rates among laboratories for both assays. The probability of detection associated with within-pen prevalence was calculated using the results from all laboratories and is shown in Figure 1. Overall, for the 100 samples from pens containing ≥1 positive pig and tested at the 6 laboratories, the mean positivity was 62% for PRRSV RNA and 61% for PRRSV antibody.
Porcine reproductive and respiratory syndrome virus (PRRSV) real-time reverse transcription polymerase chain reaction (RT-PCR) results for oral fluid samples from pens of known PRRSV prevalence.*
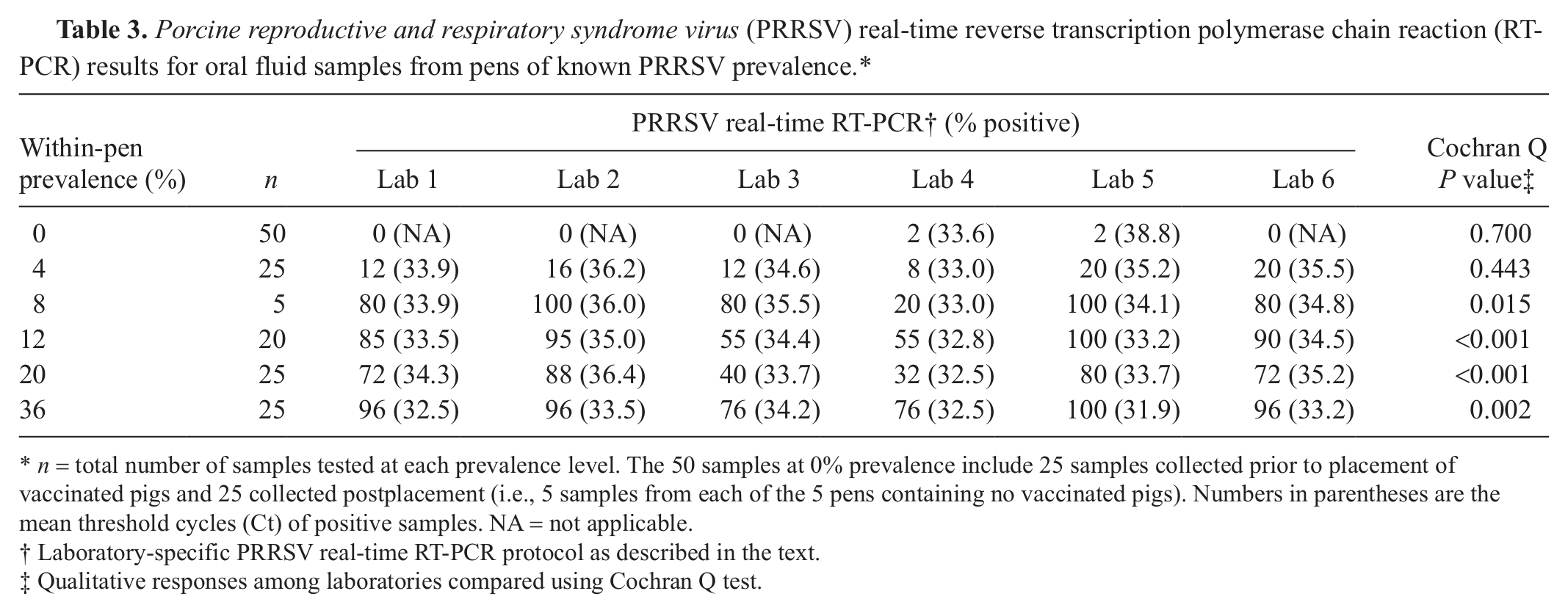
n = total number of samples tested at each prevalence level. The 50 samples at 0% prevalence include 25 samples collected prior to placement of vaccinated pigs and 25 collected postplacement (i.e., 5 samples from each of the 5 pens containing no vaccinated pigs). Numbers in parentheses are the mean threshold cycles (Ct) of positive samples. NA = not applicable.
Laboratory-specific PRRSV real-time RT-PCR protocol as described in the text.
Qualitative responses among laboratories compared using Cochran Q test.
Porcine reproductive and respiratory syndrome virus (PRRSV) enzyme-linked immunosorbent assay (ELISA) results for oral fluid samples from pens of known PRRSV prevalence.*
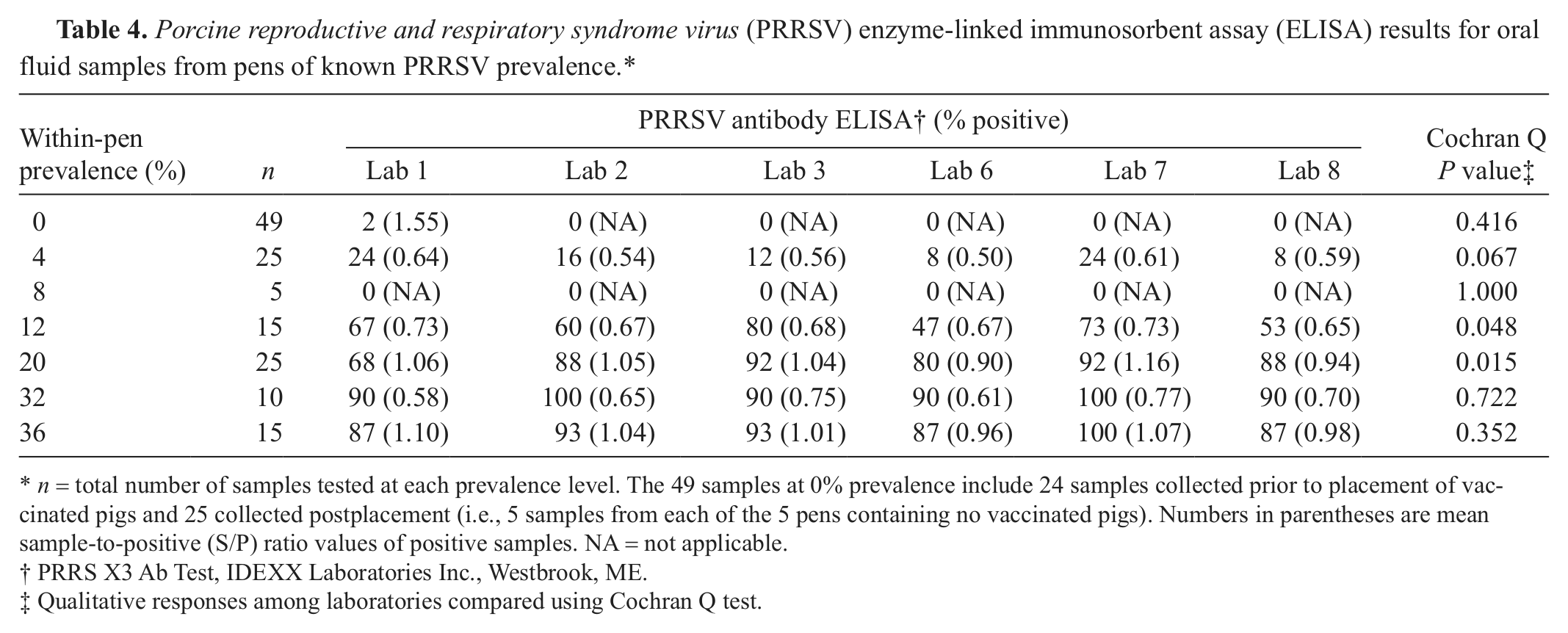
n = total number of samples tested at each prevalence level. The 49 samples at 0% prevalence include 24 samples collected prior to placement of vaccinated pigs and 25 collected postplacement (i.e., 5 samples from each of the 5 pens containing no vaccinated pigs). Numbers in parentheses are mean sample-to-positive (S/P) ratio values of positive samples. NA = not applicable.
PRRS X3 Ab Test, IDEXX Laboratories Inc., Westbrook, ME.
Qualitative responses among laboratories compared using Cochran Q test.
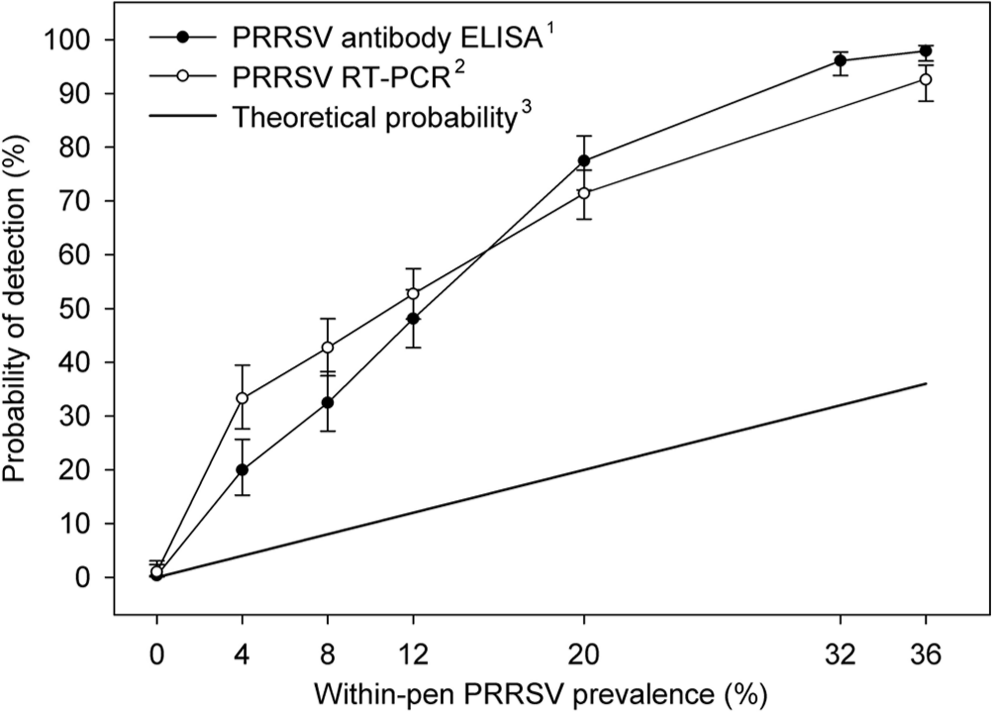
Probability of a Porcine reproductive and respiratory syndrome virus (PRRSV)–positive oral fluid sample as a function of within-pen prevalence modeled using logistic regression. The analysis was based on testing preformed at 8 laboratories (6 performed antibody assays, 6 performed nucleic acid assays). (1) PRRSV antibody enzyme-linked immunosorbent assay (ELISA): PRRS X3 Ab Test, IDEXX Laboratories, Inc. (2) PRRSV real-time reverse transcription polymerase chain reaction (RT-PCR): see text for laboratory-specific protocols. (3) Theoretical probability of identifying a positive pen by randomly selecting 1 pig and testing using a perfect test.
The results of collection order analysis on qualitative RT-PCR and ELISA results did not show statistically significant differences (P ≤ 0.05). The Cochran Q P value for RT-PCR and ELISA was 0.521 and 0.196, respectively.
Discussion
A continuous flow of accurate, real-time disease information is needed to support animal health decisions in commercial swine populations, improve the quality of field research, and provide timely information on endemic and foreign animal diseases in the national swine herd. Historically, the primary roadblock to this goal has been the expense of collecting and testing statistically appropriate numbers of blood, feces, or nasal swab specimens from individual pigs. In contrast, collecting oral fluid specimens requires less labor, is stress free for both animals and human beings, and is a sensitive method for detecting infections in populations.5,9-12 For these reasons, oral fluid sampling has been rapidly adopted by producers and veterinarians and has become routine on many farms. At the Iowa State University Veterinary Diagnostic Laboratory (Ames, IA), the specimen type “swine oral fluid” was entered into the laboratory information management system in February 2010. Thereafter, 10,329 oral fluid specimens were received for testing in 2010 and 32,517 in 2011, and >58,000 are expected in 2012.
The first oral fluid–based antibody ELISA for swine, the PRRSV oral fluid Ig ELISA, was described in 2012. 6 Prior to this, the majority of swine oral fluid specimens were tested by PCR because antibody assays for pathogens of swine had not been adapted to the oral fluid matrix. The use of oral fluid PCRs was supported by research demonstrating their application to the oral fluid matrix.1,5,9-12 Routine implementation of the PRRSV oral fluid Ig ELISA test in diagnostic laboratories was supported by extensive assessment of assay performance. Specifically, an interlaboratory study of the ELISA using 276 oral fluid samples found 97.5% agreement among 12 participating laboratories. 7
Regardless of the analytical methodology, analyte detection using pen-based oral fluid differs most significantly from individual animal sampling in that pen-based oral fluid specimens are composed of the “voluntary” contributions of individual pigs. Logically, diagnostic results are affected both by the infection status of the individuals in the pen and their interaction with the sampling device. The intent of the present study was to begin to explore the dynamics of PRRSV detection using pen-based oral fluid samples as a function of within-pen prevalence. It was not the intent of this study to compare interlaboratory performance or testing protocols; rather, testing at multiple laboratories assured realistic and robust estimates.
While the results showed that testing pen-based oral fluid samples greatly improved detection over single-animal testing, it should be recognized that the estimates were more conservative than would be expected under typical field conditions for the following reasons: 1) PRRSV modified live vaccine produces a lower level of viremia and lower antibody response than infection with wild-type PRRSV 3 ; 2) recent mixing of vaccinated and unvaccinated pigs, while necessary to precisely establish within-pen prevalence in the current study, is known to disrupt social order and normal pig behavior 13 (e.g., interaction with the rope sampling device); and 3) although testing results varied considerably among laboratories, all results were included in the analyses.
Cumulatively, the research indicates that oral fluids are an effective method to surveil for PRRSV and other swine pathogens in swine populations. Like any infant technology, the full implementation of oral fluid diagnostics faces challenges. The most obvious of these is the lack of a full complement of assays designed to function with oral fluids. In particular, validated assays for oral fluids would be welcome because of the promise of cheaper, but more effective, surveillance for diseases of major significance (e.g., Classical swine fever virus, African swine fever virus, and others). In addition, as surveillance moves away from individual animal testing, pen-based sample size and sample frequency algorithms will be needed to effectively and efficiently implement surveillance for endemic and exotic infectious agents. Seen in this context, the present study represents the beginning of establishing a statistically sound rationale for using and interpreting pen-based oral fluid diagnostic results.
Footnotes
b.
SAS version 9.2, SAS Institute Inc., Cary, NC.
c.
Ingelvac PRRS MLV, Boehringer Ingelheim Vetmedica Inc., St. Joseph, MO.
d.
Vacutainer, BD, Franklin Lakes, NJ.
e.
Web Rigging Supply Inc., Lake Barrington, IL.
f.
BL-44, Dyna-Jet Products, Overland Park, KS.
g.
PRRS X3 Ab Test, IDEXX Laboratories Inc., Westbrook, ME.
h.
Goat, anti-pig IgGFC, Bethyl Laboratories Inc., Montgomery, TX.
i.
Ambion MagMAX-96 Viral RNA Isolation kit, Life Technologies Corp., Carlsbad, CA.
j.
KingFisher 96 Thermo Fisher Scientific, Waltham, MA.
k.
VetMAX TaqMan NA and EU PRRSV Reagents, Life Technologies Corp., Carlsbad, CA.
l.
Xeno RNA (1,000 copies/μl), Life Technologies Corp., Carlsbad, CA.
m.
EZ-PRRSV MPX 4.0, Tetracore Inc., Rockville, MD.
n.
Ambion MagMAX pathogen RNA/DNA kit, Life Technologies Corp., Carlsbad, CA.
o.
MagMAX Express-96 Magnetic Particle Processor, Life Technologies Corp., Carlsbad, CA.
p.
Ag-Path PRRS PCR kit, Life Technologies Corp., Carlsbad, CA.
q.
Catalog no. AM8500, Life Technologies Corp., Carlsbad, CA.
r.
Catalog no. AM8504, Life Technologies Corp., Carlsbad, CA.
s.
Catalog no. AM8640, Life Technologies Corp., Carlsbad, CA.
t.
Catalog no. TC-9061-096, Tetracore Inc., Rockville, MD.
u.
BioSprint 96 Workstation, Qiagen Inc., Valencia, CA.
v.
MedCalc 9.2.1.0, MedCalc Software, Mariakerke, Belgium.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S. Lizano is employed by IDEXX Laboratories Inc. (Westbrook, ME); R. Rauh is employed by Tetracore Inc. (Rockville, MD); and R. Shah is employed by Life Technologies Corp. (Austin, TX). The remaining authors declare no conflicting interests with respect to their authorship or the publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported in part by the Advancement in PRRS Research Award (Boehringer Ingelheim Vetmedica Inc.) and the PRRS CAP, USDA NIFA award 2008-55620-1932. Testing costs were supported in part by participating laboratories.