
Abstract
Salmonella Pullorum and Salmonella Gallinarum are classified as biovars of Salmonella enterica subsp. enterica serovar Gallinarum. These salmonellae are the causative agents of Pullorum disease and fowl typhoid, respectively, and are widely distributed throughout the world. Although many developed countries have eradicated these diseases from commercial poultry, they are still the cause of significant economic loss in developing countries. When serovar Gallinarum is isolated, it is difficult to immediately differentiate between biovars because they are antigenically identical by serotyping. However, they cause distinct diseases with different epidemiology, and therefore it is important to differentiate them. This may be done biochemically but takes 2 to 3 days. In the present study, S. Pullorum and S. Gallinarum whole genomes were compared, and 1 genomic region of difference, which is part of the ratA gene, was chosen as a molecular marker for a polymerase chain reaction assay to differentiate rapidly between these organisms. In all, 26 strains of S. Gallinarum and 17 S. Pullorum strains were tested and successfully differentiated by the assay.
Fowl typhoid and Pullorum disease are 2 avian salmonelloses that are of major economic significance throughout the world. Fowl typhoid is caused by Salmonella enterica subsp. enterica serovar Gallinarum biovar Gallinarum (S. Gallinarum), producing high mortality rates in birds of all ages. Pullorum disease is caused by Salmonella enterica subsp. enterica serovar Gallinarum biovar Pullorum (S. Pullorum), which causes mortality mainly in young birds and is sometimes followed by persistent infection in adults with vertical transmission.1,19 Although these 2 diseases are considered under control in many developed countries, they remain a serious economic problem to livestock in countries where less effective measures of control are in place and where ambient climatic conditions may favor the environmental dissemination of these organisms.1,16 In Brazil, for example, 10 outbreaks of Pullorum disease were reported in 2004 and 83 outbreaks of fowl typhoid from 2005 to 2009 (World Organization for Animal Health: 2012, OIE-Handistatus II. Available at http://www.oie.int/hs2. Accessed October 2012). It is likely that in Brazil, as in many countries, the prevalence of these diseases is much higher than that officially reported. 1
Currently, S. Gallinarum and S. Pullorum are classified as biovars of Salmonella enterica subsp. enterica serovar Gallinarum. 15 They belong to serogroup D (both possess O antigens 1, 9, and 12 and are nonflagellated). 8 Thus, when serovar Gallinarum is isolated, it is impossible to differentiate between the 2 biovars by serotyping. 18 Because they cause distinct diseases with different epidemiology, their correct identification is necessary. 21
Differentiation by biochemical tests (e.g., gas production from glucose fermentation; fermentation of dulcitol, maltose, rhamnose, and xylose; and decarboxylation of ornithine) is not exact as some isolates may show atypical profiles.4,5,13 Furthermore, such tests are also labor intensive. In contrast, molecular tests have been applied to accelerate differentiation.9,14,18 However, they were based on molecular markers identified before the complete genomic sequences for these biovars were available. Some of the published tests require enzymatic digestion of amplicons 10 or more than 1 polymerase chain reaction (PCR). 18 In the present study, the available genomic information has been used to find a suitable molecular maker for a PCR assay to differentiate S. Gallinarum and S. Pullorum.
The available whole genome sequences of S. Pullorum (strain RKS5078, GenBank accession no. CP003047.1) and S. Gallinarum (strain 287/91, GenBank accession no. AM933173.1)7,20 were compared using an appropriate softwarea,3; 7 genomic regions of difference (RODs) were identified that might be used as targets for PCR assays (Table 1). After analyzing the contents of all RODs, ROD 4, which is part of the ratA gene (a hypothetical protein), was chosen.
Genomic location of supposedly useful targets to differentiate Salmonella enterica subsp. enterica serovar Gallinarum biovars Gallinarum and Pullorum.
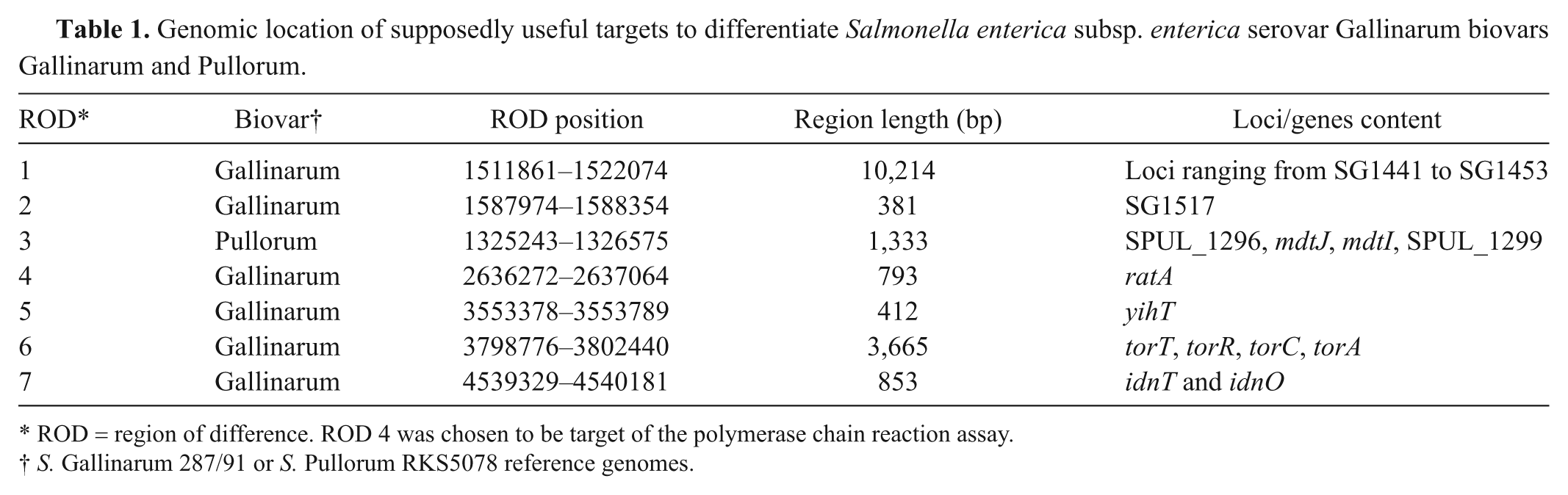
ROD = region of difference. ROD 4 was chosen to be target of the polymerase chain reaction assay.
S. Gallinarum 287/91 or S. Pullorum RKS5078 reference genomes.
Forward (5’-GACGTCGCTGCCGTCGTACC-3’) and reverse (5’-TACAGCGAACATGCGGGCGG-3’) primers targeting the ratA ROD were designed and tested, in silico. 17 Analysis showed that the primers should generate a single product that was either 1,047 bp (S. Gallinarum) or 243 bp (S. Pullorum). Sensitivity was measured by serial dilution of DNA template ranging from 30 ng to 0.3 pg.
In all, 26 S. Gallinarum and 17 S. Pullorum strains were tested. The majority of strains were isolated in different years from poultry flocks, reared in different Brazilian regions, diagnosed with fowl typhoid or Pullorum disease. These strains were isolated and identified by serotyping and biochemical tests at The National Agriculture and Livestock Laboratory (LANAGRO), which is the official laboratory of the Brazilian Ministry of Agriculture, Livestock and Supply and gives support to the surveillance of animal diseases. One S. Pullorum strain (449/87) was a British isolate. 2 Strains were removed from stock vials frozen at −80°C, inoculated in lysogeny broth (LB), b and incubated at 37°C, with shaking for 24 hr. Chromosomal DNA was extracted using a commercial kit c following the manufacturer’s instructions. The purity and concentration of genomic DNA was checked using a spectrophotometer. d Strains from American Type Culture Collection (ATCC) 9184 and ATCC 9120 were used as positive controls for S. Gallinarum and S. Pullorum, respectively.
The PCR assays used a reaction solution containing 1× buffer with KCl, e 160 μM of each deoxynucleotide triphosphate, e 1.5 mM of MgCl2, e 0.6 μM of each primer, e 0.75 U of the Taq DNA polymerase, f 1 μl of DNA template (at least 10 ng/µl), and ultra-pure water g up to 25 μl. Cycling conditions were initial denaturation at 94°C for 3 min, followed by 25 cycles at 94°C for 1 min, 63°C for 30 sec, and 72°C for 60 sec, with a final step of 72°C for 5 min. The PCR results were analyzed by electrophoresis at 4 V/cm for 60 min in a 1.5% (w/v) agarose gel stained with ethidium bromide followed by imaging.
The ratA gene had already been described as a suitable molecular marker in a previous study involving differentiation of another Salmonella serovar 11 and, for this reason, the ROD located at this gene was chosen as a marker in the present study. The PCR assay based on ratA was tested in 26 strains of S. Gallinarum and 17 strains of S. Pullorum (previously differentiated by biochemical tests) and proved to be able to differentiate all of them. Moreover, the test was able to detect down to 30 pg of DNA. The time savings and practicality are clear advantages over the current biochemical tests.
In the 2000s, other DNA-based diagnosis tests have been developed to differentiate these biovars, some of which require restriction fragment length polymorphism analysis after conventional PCR.10,14 Although such tests have facilitated the differentiation of S. Pullorum from S. Gallinarum, the cost of additional enzymes and the requirement of additional steps are obvious disadvantages when compared against the assay presented herein. Previous studies have used an allele-specific PCR assay based on polymorphism of the rfbS gene to differentiate the 2 biovars.6,18 For such tests, a given DNA sample must be tested twice using different primer combinations. Moreover, the absence of amplicons is regarded as a positive result, which itself may be confounded by the risk of false negatives as a result of technical failures. In contrast, the PCR assay described herein generates amplicons of distinct sizes for each biovar (Fig. 1), thus avoiding this potential problem.
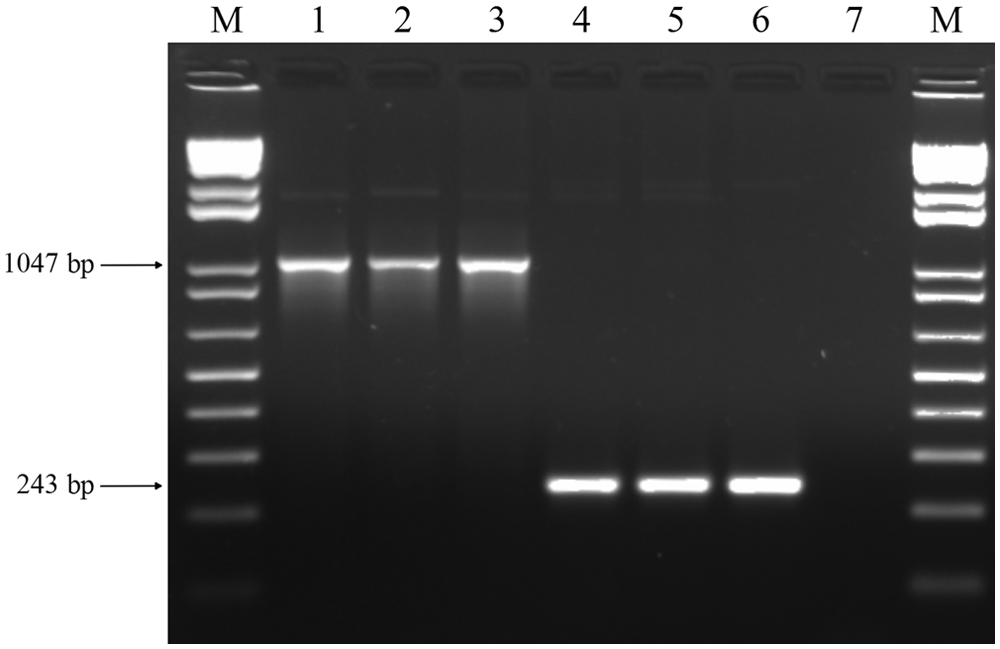
Polymerase chain reaction products from Salmonella enterica subsp. enterica serovar Gallinarum biovars Gallinarum and Pullorum strains using the ratA differential primers. Lane 1: S. Gallinarum, strain ATCC 9184; lanes 2, 3: S. Gallinarum field isolates from clinical avian samples; lane 4: S. Pullorum, strain ATCC 9120; lanes 5, 6: S. Pullorum field isolates; lane 7: negative control; lane M: 1-kb DNA ladder. e
In 2011, a duplex PCR assay was developed based on an 11-bp deletion in the glgC gene (a pseudogene in S. Gallinarum) and a 4-bp deletion in speC (a pseudogene in both biovars).9,20 In bacterial genomes, pseudogenes are continually created from ongoing mutational processes and are subject to degradation and removal by further accumulation of mutations. Their retention time seems to be extremely short and, even in very closely related bacteria, they tend to be deleted at a relatively rapid rate. 12 The 793-bp difference between the biovars used in the current study occurs in ratA, a gene which, from the genome annotation, is not a pseudogene in S. Gallinarum or S. Pullorum. In addition, no premature stop codons were noticed in the open reading frames of ratA in either biovar. The ratA gene has also not been found to be a pseudogene in any other Salmonella serovar examined to date.
The ROD located at ratA is thus more suitable for differentiation between S. Gallinarum and S. Pullorum than markers used previously. Although the PCR assay based on this gene may show amplicon in other Salmonella enterica subsp. enterica serovars, results suggest that the assay is a powerful tool for differentiating these 2 biovars when performed from isolated colonies of the serovar Gallinarum.
Footnotes
Acknowledgements
The authors are grateful to Márcia Jusi, Ketherson Silva, and the whole group at the Laboratory of Immunoparasitology from the Faculty of Agriculture and Veterinary Sciences, Universidade Estadual Paulista for technical assistance. Furthermore, the authors would like to thank the National Agriculture and Livestock Laboratory (LANAGRO) for providing all of the Brazilian S. Gallinarum and S. Pullorum strains and to Dr. Paul A. Barrow for providing the British S. Pullorum strain used in this study.
b.
Difco Laboratories Inc., Sparks, MD.
c.
QIAamp DNA Mini Kit, Qiagen GmBH, Hilden, Germany.
d.
NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific Inc., Waltham, MA.
e.
Invitrogen Corp., Carlsbad, CA.
f.
Fermentas, Thermo Fisher Scientific Inc., Waltham, MA.
g.
Sigma-Aldrich, St. Louis, MO.
Declaration of conflicting interests
The author(s) have no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the São Paulo Research Foundation (FAPESP), the Coordination of Improvement of Higher Education Personnel (CAPES), and the National Council of Scientific and Technological Development (CNPq), Brazil.