
Abstract
Scrapie is a transmissible spongiform encephalopathy of sheep and goats and is associated with the deposition of an abnormal isoform of prion protein (PrPsc). This isoform presents an altered conformation that leads to its aggregation in the host’s central nervous and lymphoreticular systems. A predisposition to the prion-agent infection can be influenced by specific genotypes that are related to polymorphisms in the ovine prnp gene. The most characterized polymorphisms occur at codons 136, 154, and 171, with genotype VRQ being the most susceptible and ARR the most resistant. In the current study, a real-time quantitative polymerase chain reaction (qPCR) technique based on allele-specific TaqMan probes was developed to identify single nucleotide polymorphisms in the prnp gene from Brazilian herds. Specific primers and TaqMan probes were designed for all 3 codons of interest. Samples from a total of 142 animals were analyzed by qPCR, followed by DNA sequencing of the amplicons. All of the genotypes determined by qPCR were in agreement with the data determined by DNA sequencing. In all 3 of the analyzed breeds, the majority of the animals were AA homozygous for the 136 codon. The most frequent genotype for codon 154 was RR, and genotypes QQ and QR were the most frequent for codon 171. The results are discussed in relation to establishing scrapie control measures and breeding programs for Brazilian herds.
Scrapie, a neurodegenerative disease affecting sheep and goats, is one of several transmissible spongiform encephalopathies or prion diseases. The disease is characterized by the accumulation of an abnormal isoform (PrPsc) of a host-encoded cellular prion protein PrP c in the central nervous system. 13 Although scrapie is an infectious disease, the susceptibility of sheep is strongly influenced by polymorphisms of the prion protein gene ( prnp). 4 The combined detection of polymorphisms at codons 136, 154, and 171 suggests that VRQ (valine, arginine, glutamine) and ARR (alanine, arginine, arginine) are antagonistic in determining the susceptibility to the disease: VRQ is associated with a high incidence of natural scrapie, and ARR is associated with a low incidence of natural scrapie. 6
Programs for selecting sheep with PRNP genotypes that appear to confer resistance to the development of classical scrapie has been used for several years in an attempt to eradicate the disease. 14 However, in Brazil, there is no selection program in operation related to scrapie, even though research in sheep genetics and breeding has increased significantly in recent years. Such studies include research on the characterization, breeding, and crossing of sheep using new technologies that incorporate both classical quantitative and molecular genetics. 19 Since 2007, some studies have analyzed the PRNP genotypes in Brazilian sheep herds.2,15,17,21,22 However, all of this research is based on sequencing and/or restriction fragment length polymorphism (RFLP) and has been applied to a limited number of animals. Several PRNP genotyping methodologies for detecting single nucleotide polymorphisms (SNPs) have been described, including sequencing, RFLP, mass spectrometry, quantitative polymerase chain reaction (qPCR), and amplicon melting temperature analysis.1,3,12,18
Direct sequencing accurately determines the genotype of the samples with respect to the SNPs in codons 136, 154, and 171 and will reliably identify any additional polymorphisms in neighboring codons. However, SNP genotyping technologies, such as sequencing and RFLP, are costly for the analysis of a large number of samples. 9 TaqMan a SNP genotyping is a well-proven technique and has been successfully used to determine PRNP genotypes in sheep. 24 Moreover, a careful design of the probes used should prevent cross-reactions with a mismatched probe and the generation of a nonspecific signal. 14 The main benefit of sequence-specific detection is that it allows the unambiguous detection of target sequences without the contaminating signals that arise from primer dimers and other nonspecific PCR events. This is due to the low likelihood that a nonspecific PCR will generate a hybridization site for an oligonucleotide probe to bind and generate a signal.10,11,25 Furthermore, TaqMan probes are the most widely used technology for qPCR. The aim of the present study was to develop and apply a qPCR TaqMan assay for SNP genotyping that would reliably identify alleles A/V in the 136 codon, alleles R/H in the 154 codon, and alleles Q/R/H in the 171 codon of the sheep prnp gene.
Whole peripheral blood samples were collected using ethylenediamine tetra-acetic acid evacuated blood collection tubes b from 142 animals belonging to the Suffolk breed (100 animals), Dorper/White Dorper breed (19 animals), and Santa Inês breed (23 animals). All of the animals were from herds originating in the southern and southeastern regions of Brazil. The genomic DNA was extracted from 500 µl of whole blood using a commercially available kit, c following the manufacturer’s instructions. The DNA was recovered in 200 µl of elution buffer and stored under refrigeration for further analyses. Every genomic DNA sample was quantified using a spectrophotometer d and diluted to 20 ng/μl final concentration for use in the PCR reactions.
Each sequence-specific probe, representing alternate alleles of the prnp gene, was designed with a different fluorescent label, and both were combined in a reaction with the same set of primers. The probes and primers were designed for the Ovis aries prnp gene (GenBank accession no. M31313) using commercial software. e The primers and probes sequences used for the allelic discrimination assays at the 136 codon were: forward (5′-GGGCCTTGGTGGCT ACATG-3′), reverse (5′-TCCTCATAGTCATTGCCAAAA TGTAT-3′), 136A probe (5′-6FAM-TGGGAAGTGCCA TGAG-3′), and 136V probe (5′-VIC-TGGGAAGTGTCA TGAG-3′). Primers and probes used for codon 154 were: forward (5′-GGCCTCTTATACATTTTGGCAATG-3′), reverse (5′-ATCCACTGGTCTGTAGTACACTTGGT-3′), 154R probe (5′-6FAM-ACCGTTACTATCGTGAAA-3′), and 154H (5′-VIC- ACCGTTACTATCATGAAA-3′). Primers and probes used for codon 171 were: forward (5′-GTTACCCCAACCAA GTGTACTACAGA-3′), reverse (5′-TGTTGACACAGTCA TGCACAAAG-3′), 171Q probe (5′-6FAM-CAGTGGATCA GTATAGTAA-3′), 171R probe (5′-VIC-CAGTGGATCGG TATAG-3′), and 171H probe (5′-NED-CAGTGGATCATTA TAG-3′). All probes contained MGBNFQ (minor groove binding nonfluorescent quencher) at the 3′-end. The PCR conditions were previously optimized for the temperature and concentrations of the probe, primer, and magnesium chloride. All of the optimizations were performed using samples of known genotypes. The reaction efficiencies were determined using calibration curves for each set of primers and probes. The primers were tested at 10, 15, 20, and 25 pmol, and the probes were tested at 120, 200, and 300 nM. The hybridization temperatures were tested in a gradient ranging from 58ºC to 66ºC (2ºC step). The magnesium chloride concentration ranged from 1 to 3 mM. For codon 171, a triplex qPCR was tested that included probes 171Q, 171R, and 171H without success; reactions could only be optimized for 2 duplex reactions: 171Q/171R and 171Q/171H.
The PCR reactions contained 20 pmol each primer, 200 μM of deoxyribonucleotide triphosphate (dNTPs), 1× Taq buffer, f 1 U Taq DNA polymerase, f and 1 μl of genomic DNA (20 ng) in a final volume of 25 μl. The concentrations of magnesium chloride and probes, and the hybridization temperatures, varied in the 4 reaction sets. The reactions were performed using a real-time PCR system, e and the PCR reaction for each sample was performed in duplicate. A negative control in duplicate was included for every 96-well plate run.
The PCR reactions for obtaining amplicons for sequencing were performed using a forward primer flanking the 136 codon position and a reverse primer flanking the 171 codon position, generating a 245-bp amplicon. 16 The PCR reactions contained 15 pmol of each primer, 1.5 mM of MgCl2, 200 μM of dNTPs, 1× Taq buffer, f 1 U of Taq DNA polymerase, f and 1 μl of genomic DNA in a final volume of 25 µl. The PCR reactions were performed using a automated DNA thermal cycler e with the following parameters: 95ºC for 5 min; followed by 35 cycles at 95ºC for 30 sec, 58ºC for 30 sec, and 72ºC for 30 sec; and a final step at 72ºC for 10 min.
The PCR products were purified using a commercial kit f and quantified using a fluorescence quantification system f according to the manufacturer’s instructions. The sequencing of the PCR products was performed using Sanger sequencing. e Each sample was sequenced independently using both forward and reverse primers until a Phred quality score of at least 20 was obtained for each individual base in the consensus sequence.7,8 The resulting chromatograms were analyzed using the Staden package version 1.7.0 program 23 and novoSNP version 3.0.1. 26
Initially, the standardization of the qPCR was accomplished using samples that were previously sequenced and with a known genotype. For each set of reactions, different concentrations of the primers, probes, and magnesium chloride were tested, as were the temperature gradients. Four different reaction sets were established for SNP genotyping the variations at codons 136 (A/V), 154 (R/H), and 171 (Q/R and Q/H). The optimum hybridization temperatures were 66ºC (136 codon reaction set), 60ºC (154 codon reaction set), and 62ºC (both 171 codon reaction sets). All of the reaction sets contained 1 mM of MgCl2, except the 136 reaction set for which a concentration below 3 mM led to a low specificity. The optimum probe concentration was determined to be 120 nM, except for probes 136V and 171H for which a concentration below 300 nM produced a very weak fluorescence signal.
A representative amplification plot for probes 171Q and 171R is shown in Figure 1. Both the amplification curves and the ΔRn at the final cycle are diverse enough to allow differentiation of the 3 possible genotypes. For both of the probes, the ΔRn values for the samples from heterozygote animals are almost half the value when compared to those from the homozygous animals. In addition, the curves for the duplicates of each sample are almost identical, and the samples from homozygous animals have a ΔRn close to zero with the mismatch probe.
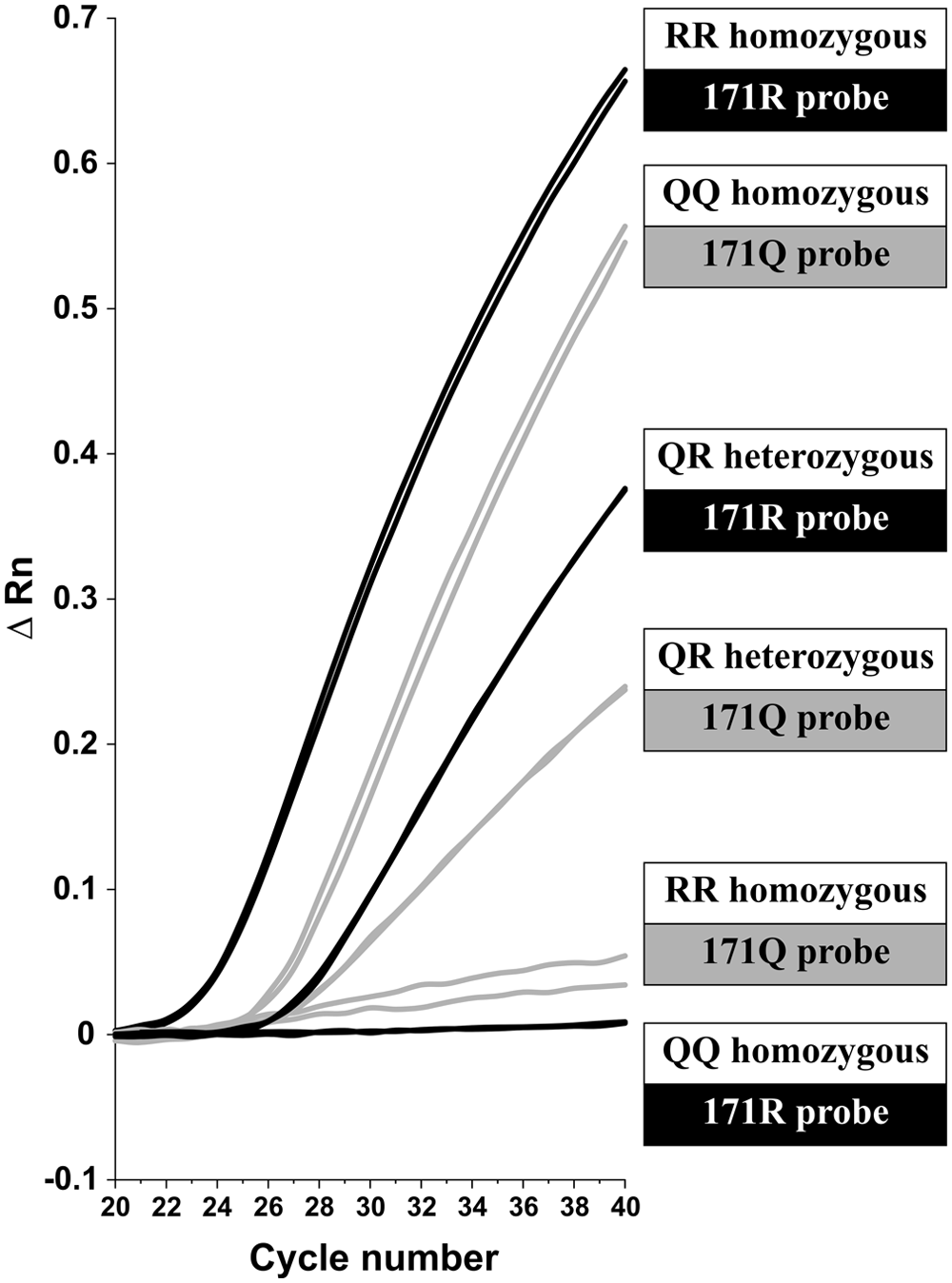
Representative amplification plots for quantitative polymerase chain reaction (qPCR) using TaqMan probes 171Q and 171R. The DNA from 3 animals with genotypes RR, QQ, and QR for codon 171 was subjected to a duplex qPCR using probes 171Q and 171R. Each sample was analyzed in a duplicate reaction.
To establish the cutoffs to assign the possible genotypes to each sample, the mean ΔRn values for each sample duplicate obtained for each probe pair were compared. In Figure 2, the plot obtained for probes 171Q and 171R is shown applied to 138 of the samples. All of the samples from the homozygous animals (QQ and RR) presented very low fluorescence with the mismatch probe, and the samples from the heterozygous animals presented intermediate fluorescence with both of the probes. The mean ΔRn values (endpoint) for qPCR using TaqMan probes 171Q were 0.62 (± 0.19), 0.32 (± 0.07), and 0.02 (± 0.02) for genotypes QQ, QR, and RR, respectively. The mean ΔRn values (endpoint) for qPCR using TaqMan probes 171R were 0.74 (± 0.19), 0.44 (± 0.11) and 0.00 (± 0.01) for genotypes RR, QR, and QQ, respectively.
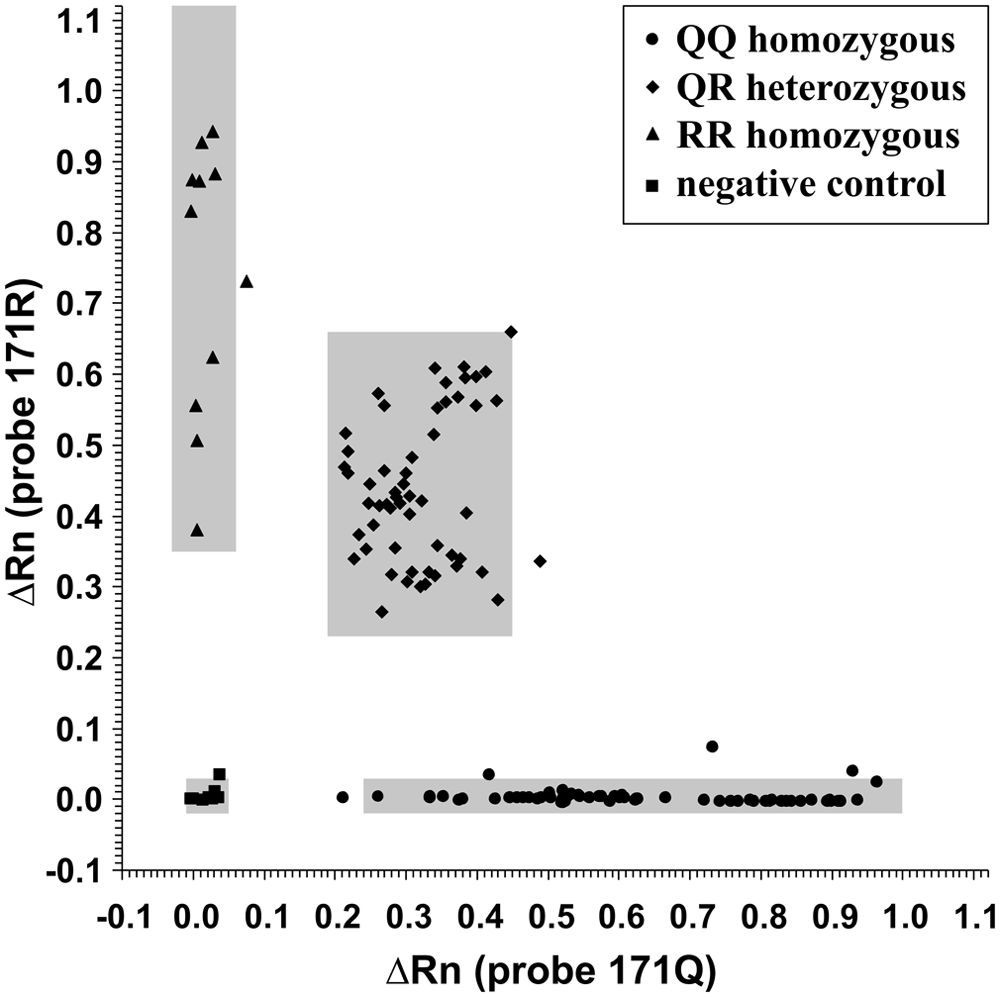
ΔRn values for quantitative polymerase chain reaction using TaqMan probes 171Q and 171R. Each point represents the mean ΔRn value for the duplicates. The gray areas indicate the standard deviation.
The amplicons from all 142 of the samples were sequenced to validate the real-time PCR results, and no discrepancies were found. Duplicate independent amplicons were obtained and sequenced for those animals with genotypes considered to be of a low frequency (136AV, 136VV, 154RH, 171RR, 171QH, and 171HH).
A previous study concluded that the real-time PCR method is superior to RFLP because of the increased repeatability of the assays, the simple one-step assay protocol, and the clarity of the results obtained compared to RFLP in which an incomplete digestion of the PCR products can produce ambiguous results. 10 In the current study, the results showed that the assay of SNP genotyping using real-time PCR for the detection of SNPs in the prion protein gene is efficient due to its accuracy, rapidity, and the specificity of the results; furthermore, the technique did not require post-PCR manipulations.
The genotype distribution for SNPs at the prnp gene found among the analyzed herds is shown in Figure 3. All known allelic forms at the 136 codon were found in the present study. For the 154 codon, the homozygous allele for histidine (HH) was not found and, for codon 171, none of the animals presented the heterozygous allele RH.
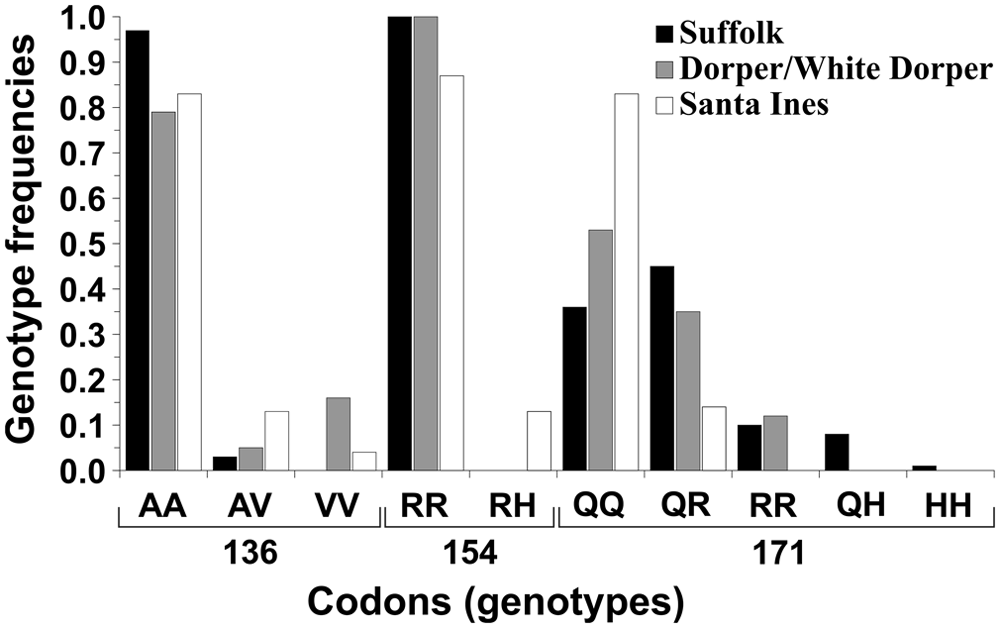
Genotype frequencies for single nucleotide polymorphisms at codons 136, 154, and 171 of the prnp gene. Only the genotypes found among the analyzed Suffolk, Dorper/White Dorper, and Santa Inês sheep are shown.
The allele frequencies estimated for the Suffolk breed found in the current study at codons 136 and 154 were similar to the frequencies reported by other studies analyzing herds from Brazil (Sotomaior CS: 2007, Polimorfismo do gene da proteína prion celular (PrPC) e a susceptibilidade/resistência ao scrapie em ovinos no Estado do Paraná [Polymorphism of the cellular prion protein gene (PrPC) and susceptibility/resistance to scrapie in sheep in the State of Paraná]. Doctoral thesis in Animal Health, Faculty of Veterinary Sciences, Universidade Federal do Paraná, Curitiba, PR, Brazil).2,21 However, at codon 171, a higher variety of genotypes was found when compared to these previous studies (Fig. 3). The present study found Q171R to be the most frequent (44.5%) genotype in Suffolk animals, in accordance with previous studies including German and Irish Suffolk breeds (40–42%).5,15,20 However, some other previous studies in Brazil found higher frequencies for homozygosity at 171Q.21,22
Studies including Dorper and White Dorper herds from Brazil and elsewhere in the world showed genotype frequencies similar at codon 154 (Fig. 3).12,15,22 For codon 136, it was found that allele V was homozygous with a 15.8% frequency and that genotype A136V had a 5.3% frequency (Fig. 3). Previous studies reported a higher frequency in A136V (20–25%).15,22 Homozygous genotype 171Q was the most frequent in Dorper and White Dorper (52.9%; Fig. 3), but other authors found frequencies as high as 75–80%.15,22
For the Santa Inês breed, genotype frequencies at codon 136 and 154 were found to be quite similar to previous works with Brazilian herds of approximately the same size, varying from 0.82 to 0.89 (Fig. 3).17,22 However, another study with different herds also from Brazil, but with a larger number of animals (421), found frequencies as low as 0.68 for the homozygous 154R genotype. 15 Finally, at codon 171, frequencies of 0.83 and 0.17 were found for genotypes 171Q and Q171R, respectively, differing from previous studies with Brazilian herds that found frequencies of 0.60 and 0.40 for these genotypes.15,22
Taken together, the results of the current study indicate that the herds of Santa Inês and Dorper/White Dorper present a high frequency of animals with scrapie-susceptible genotypes. The extension of the present work to include a larger number of animals from different herds, regions, and breeds from Brazil will allow for a clearer overview of the strategies that should be applied in order to reduce the harmful impact of classical scrapie on the sheep production system in Brazil. The authors plan to broaden the study in order to analyze other polymorphisms in the prnp gene related to atypical scrapie, such as the polymorphism at the 141 codon.
Footnotes
Acknowledgements
The authors would like to thank Juliano de Souza Leal and Gabriel Laizola Frainer Correa for technical assistance in collecting the samples.
a.
F. Hoffmann-La Roche Ltd., Basel, Switzerland.
b.
Vacutainer, BD, Franklin Lakes, NJ.
c.
QIAamp DNA blood mini kit, Qiagen Biotecnologia Brasil Ltda, São Paulo, Brazil.
d.
GeneQuant 1300, GE Healthcare Technologies, Piscataway, NJ.
e.
Primer Express version 3.0 software, StepOnePlus real-time PCR system, Veriti automated DNA thermal cycler, BigDye terminator cycle sequencing kit version 3.1 (in an ABI PRISM 3130 Genetic Analyzer); Applied Biosystems, Life Technologies do Brasil, São Paulo, Brazil.
f.
Platinum buffers, PureLink PCR purification kit, Qubit fluorescence quantification system; Invitrogen Brasil Ltda, Sao Paulo, Brazil.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Caroline Pinto de Andrade was supported by a fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; Proc. 578226/2008-1), and financial support was provided by CNPq (Proc. 505917/2008-4).