
Abstract
Classical scrapie disease is a transmissible spongiform encephalopathy of sheep that is enzootic in the United States. Susceptibility of sheep to classical scrapie is linked to single nucleotide polymorphisms in the prion protein gene (PRNP), forming the basis for genetic testing strategies used by national efforts to eradicate scrapie. Such efforts are occasionally hampered by inconclusive results stemming from the detection of “complex” genotypes. Naturally occurring cases of ovine chimerism are thought to account for some of these instances. In the current report, 4 naturally occurring ovine chimeras are documented through cytogenetic and molecular analyses. All 4 of these sheep had chimeric cells circulating in their blood. Blood and alternate tissue samples of ear punch and hair bulbs from one of these chimeras was submitted in batch with similar samples from control sheep for routine scrapie genetic relative susceptibility testing. A complex PRNP genotype was detected in the blood of the chimeric female but not in the alternate tissue samples or in the control sheep samples. The results demonstrate that naturally occurring blood chimerism can confound current testing efforts. The potential impacts of undetected chimeras on current scrapie eradication efforts are discussed.
Introduction
The “chimaera” of Homer's Iliad is a mythical creature composed of parts from different animals. 5 The modern clinical usage of “chimera” denotes an animal whose cellular composition persistently includes cells derived from a nonidentical, or dizygotic, twin. In ruminants, natural chimerism commonly results from an exchange of stem cells made possible by the early formation of placental vascular anastomosis between twins. 28 Though not as outwardly obvious as the mythical creature, chimeric cells are nonetheless genetically disposed to express the protein isoforms native to the twin sibling. Thus, abnormally complex blood types, a result of immune tolerance to antigens expressed by erythrocytes, have long been recognized as signifying the presence of twin-derived erythrocytes circulating in the blood of chimeras. 36
Scrapie is a slowly progressive, fatal neurologic disease of sheep that is critically associated with conversion of the host's cellular prion protein (PrP c ) to a misfolded form (PrPSc). 31 Amino acid substitutions within PrP c naturally occur as a result of single nucleotide polymorphisms in its gene, PRNP. Of particular importance to scrapie disease are polymorphisms encoding valine or alanine at codon 136 (V136 or A136, respectively), arginine or histidine at codon 154 (R154 or H154, respectively), and glutamine, arginine, histidine, or lysine at codon 171 (Q171, R171, H171, or K171, respectively). 12 Genetic relative susceptibility testing categorizes individuals into disease risk groups associated with the inherited alleles. Of current importance to relative susceptibility for classical scrapie disease in sheep are the alleles A136R154R171 (hereafter, ARR; which is associated with resistance), and A136R154Q171 (ARQ) and V136R154Q171 (VRQ), which are both associated with susceptibility. 20
National programs, including the U.S. Scrapie Eradication and Surveillance programs, aim to control scrapie disease by increasing the frequency of stock genetically resistant to disease through PRNP genotype determination (U.S. Department of Agriculture: 2008, National Scrapie Surveillance Plan. Available at http://www.aphis.usda.gov/vs/nahss/sheep/national_scrapie_surveillance_plan_08192008.pdf. Accessed on January 20, 2009). 12 Consideration of the PRNP genotype significantly affects the valuation of breeding stock, indicating a recognition of the importance of such national programs to the sheep industry. 15 Genetic testing is conveniently performed on blood samples, but inconclusive results arising from the detection of “complex” PRNP genotypes have been reported (<0.1% of U.K. samples). 13 Such genotype complexity has been attributed to sample contamination and to naturally occurring ovine chimeras. 14,18,26,34 However, many chimeras may be going undetected by current efforts given the reported rates of 3–5% chimeras among co-siblings in some breeds of sheep. 17,23,25,37 In the current report, 4 cases of naturally occurring ovine chimeras are characterized, and a confounding effect on current commercially contracted scrapie genetic relative susceptibility testing in the United States is demonstrated. Testing methodologies and the implications of natural ovine chimerism on genetic testing and scrapie eradication efforts are discussed.
Material and methods
Animals
Chimera case 1 consisted of a black-faced female sheep born twin to a male in the winter of 2005 on a commercial U.S. sheep operation. At approximately 18 months of age, results from genetic relative susceptibility testing for classical scrapie by a U.S. Department of Agriculture (USDA)-contract laboratory were reported as “inconclusive” after 2 independent blood sample submissions. A third blood sample was drawn at 20 months of age and submitted to the National Veterinary Services Laboratories (NVSL), a which subsequently reported that the sample had either been contaminated with DNA from another sheep or had been obtained from a chimera. The female was acquired by the Animal Disease Research Unit (ADRU) b for further investigation and was approximately 2.5 years of age at the time samples were collected for the present study.
Chimera cases 2 and 3 consisted of female lambs reportedly born twin to one another in a commercial flock. Blood samples collected at 2 months of age were submitted to a USDA-contract laboratory for standard genetic susceptibility testing, the results of which were ultimately reported back for both animals as “inconclusive.” DNA extracts from these samples were kindly provided by NVSL for the present study.
Chimera case 4 consisted of a female sheep for which no lambing history is available. A blood sample had been previously identified by NVSL as having a complex PRNP genotype. The samples provided by NVSL included frozen whole blood and a frozen ear specimen collected post-euthanasia.
Control samples were obtained from 9 mature sheep maintained by the ADRU. These animals were of the following PRNP genotypes 12 as determined by polymerase chain reaction (PCR) sequencing (described below): ARR/ARR, 2 females and 1 male; ARQ/ARQ, 3 females and 1 male; ARQ/ARR, 1 female; ARR/VRQ, 1 female.
Collection of samples
The care and use of ADRU sheep were approved by the Institutional Animal Care and Use Committee of Washington State University. All blood samples were collected via jugular venipuncture directly into prelabeled tubes. c Skin samples were collected from the ear margin using a commercially available tissue collection system d as described by the manufacturer. Hair bulb samples were collected by plucking primary (guard) hairs from either the skin-hoof margin of a forelimb or, as in chimera case 4, from the margin of the pinna using a hemostat wiped clean with alcohol before each use.
Cell cultures, chromosome preparations, and chromosome analysis
Conducted at the Molecular Cytogenetics Laboratory e (TR, PJD), metaphase chromosome preparations were obtained from short-term peripheral blood leukocyte cultures using a standard protocol. 32 In brief, 1 ml of whole blood collected into sterile glass tubes containing sodium-heparin c was added to 9 ml of culture medium containing RPMI (Roswell Park Memorial Institute)-1640 medium with GlutaMAX and 25 mmol HEPES (N-2-hydroxyethylpiperazone-N-2-ethanesulfonic acid) buffer, f 30% fetal bovine serum, g 1.4% antibiotic-antimycotic solution, f and 1% pokeweed mitogen (lectin from Phytolacca americana). h The cells were cultured at 37°C for 72 hr, and metaphases were arrested by treating cultures with demecolcine solution h (0.1 μ/ml) for 1 hr, followed by a 30-min treatment with Optimal Hypotonic Solution. i The cells were fixed in methanol/glacial acetic acid (3:1), dropped into precleaned glass slides, and then air dried. Chromosomes were stained with 5% Giemsa solution h in 0.07 mol Sørensen buffer, pH 6.8. C-banding was performed according to a previous study 4 by treating slides in 0.2 N (normal) HCl for 30 min at room temperature, in 2.5% Ba(OH)2 for 10 min at 60°C, and in 2X saline–sodium citrate (SSC) for 15 min at 60°C, followed by staining in 5% Giemsa solution for 1 hr. Chromosome preparations were viewed using a motorized microscope. j A total of 75 cells were captured and analyzed using commercial karyotyping system software. k Sheep chromosomes were arranged into karyotype according to ISCNDB2000. 9
PCR amplification and sequencing of selected genes
Information about the PCR primer pairs named below is provided in Table 1. As performed at the Molecular Cytogenetics Laboratory, DNA was extracted from peripheral blood of chimera case 1, a control male, and a control female following standard protocols. 6 The ovine androgen receptor (AR) was amplified using equine AR primers (AR_FP, AR_RP). The ovine Y-linked male sex determination gene (SRY) was amplified using bovine SRY primers (SRY_FP1, SRY_RP1). PCR reactions were carried out in a 10-μl volume with 0.25 U of Taq polymerase, 1 50 mmol KCl, 10 mmol Tris-HCl (pH 8.4), 0.2 mmol 2′-deoxyadenosine-5′-triphosphate (dATP), 2′-deoxycytidine-5′-triphosphate (dCTP), 2′-deoxythymidine-5′-triphosphate (dTTP), and 2′-deoxyguanosine-5′-triphos-phate (dGTP), 0.3 μmol of each primer, and 50 ng of DNA template. At the ADRU, DNA was extracted from samples of blood, tissue, and hair bulbs using a commercial kit m with the following changes to the manufacturer's instructions: 8 hairs were used for DNA extraction from hair bulbs, and residual ethanol was removed from blood extract pellets by allowing the inverted tube to dry overnight. In these samples, primers SRY_FP2 and SRY_RP2 were used to detect ovine SRY as described elsewhere. 3 PCR products were resolved on 1.5% or 2% agarose gels containing ethidium bromide.
Polymerase chain reaction (PCR) amplification and sequencing primers used for ovine genes in the current study.*
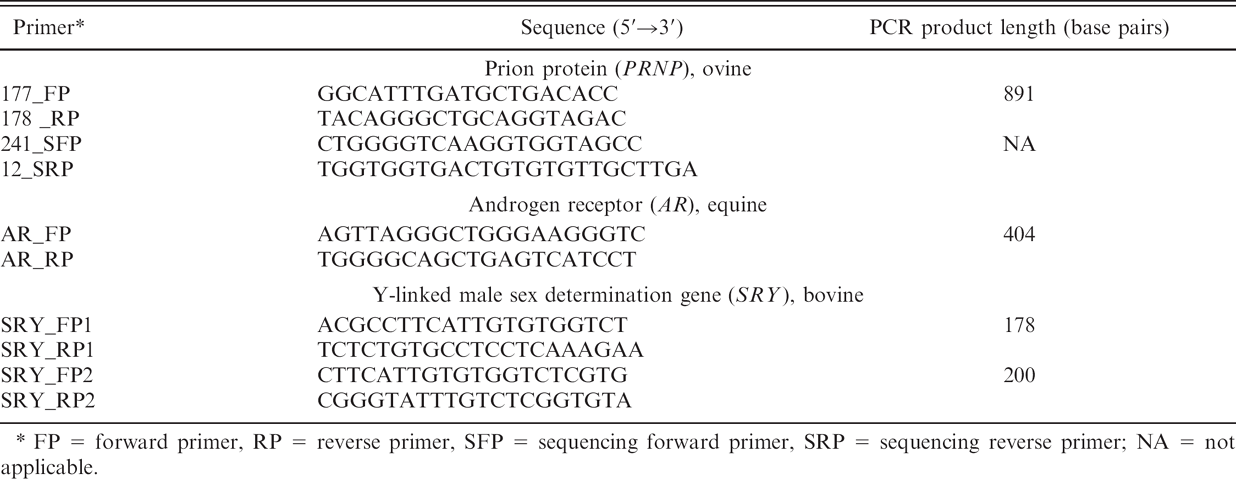
FP = forward primer, RP = reverse primer, SFP = sequencing forward primer, SRP = sequencing reverse primer; NA = not applicable.
PRNP sequencing was performed at the ADRU. PCR amplification was carried out using a commercial kit, n primers 177_FP and 178_RP, and standard buffer conditions with 2.5 mmol MgCl2, and a final volume of 50 μl. PCR conditions were 95°C for 5 min; 95°C for 30 sec, 62°C for 30 sec, 72°C for 59 sec for 30 cycles; and a final extension at 72°C for 7 min. PCR products were purified o to remove each unincorporated deoxyribonucleotide tri-phosphate (dNTP) and primer, and then sequenced p using primers 241_SFP and 12_SRP. PCR sequencing conditions were 96°C for 1 min; and 96°C for 10 sec, 50°C for 5 sec, and 60°C for 4 min for 25 cycles. Relevant PRNP genotypes are identified q in the current study as the 3 amino acids deduced from the open reading frame at codons 136, 154, and 171.
Genetic susceptibility testing was carried out by commercial laboratories certified by the USDA to conduct testing for the U.S. Scrapie Eradication Program. Each laboratory used proprietary techniques for PRNP genotype determination at codon 171.
Results
Chimera case 1
As conducted at the Molecular Cytogenetics Laboratory, chimerism in this adult female was demonstrated by the presence of 2 diploid blood cell populations: out of 75 cultured leukocytes, 42 (56%) had a normal female chromosome complement (54, XX; Fig. 1A), whereas 33 (44%) had a normal male chromosome complement (54, XY; Fig. 1B). Correct identification of X and Y chromosomes was confirmed through C-banding analysis (data not shown). The presence of the Y chromosome was also detected by PCR amplification of the Y-linked gene, SRY (Fig. 1C, lane 1). As expected, an SRY band was also detected in the blood of a male control sheep (Fig. 1C, lane 2) but not in the blood of a female control sheep (Fig. 1C, lane 3). Used as a positive control for DNA extract, an amplicon band for the X-linked AR was detected in blood samples from all 3 animals (Fig. 1C, respective lanes 5–7).
Samples of blood, ear punch, and hair bulbs obtained from chimera case 1 and control sheep were similarly probed by the ADRU for the presence of SRY; in this multiplex assay, a PRNP band served as an internal PCR reaction control in lanes containing DNA extract. As shown in Figure 2A, an SRY band was detected in each of the sample lanes for the male control sheep but not in sample lanes for the female control sheep or in reagent control lanes (Fig. 2A). From chimera case 1 (Fig. 2A, female chimera lanes), an SRY band was detected in the samples of blood and ear punch but not in a sample of hair bulbs. Commercial microsatellite analysis r was also conducted on a sample of blood from chimera case 1, the results of which are presented in Table 2 (chimera case 1). In summary, 1 allele for each of the 5 microsatellites, 2 alleles for each of the 8 microsatellites, and an “extra” allele (3 total alleles; boldfaced type) for each of the 2 microsatellites were detected.
The PRNP sequencing chromatograms obtained from blood, ear punch, and hair bulb samples of chimera case 1 (Fig. 2B) were essentially identical at codons 136 and 154, showing single nucleotide peaks at each codon with corresponding amino acid deductions of A136 and R154. The polymorphism detected at codon 171 shows the typical appearance of equivalent middle position adenine (A) and guanidine (G) nucleotide peaks in the ear punch and hair bulb samples; corresponding amino acids are QR171. This polymorphism was also detected in the blood sample, but in comparison to that observed in the samples of ear punch and hair bulbs, the middle position A nucleotide peak was substantially diminished relative to the G nucleotide peak (Fig. 2B, green arrow). Relevant nucleotide background peaks were not evident in this vicinity of the chromatogram. This minor-A-to-major-G nucleotide (“imbalanced”) appearance at codon 171 was consistently replicated after resampling this female sheep's blood and in all forward and reverse sequencing primer chromatograms (data not shown).
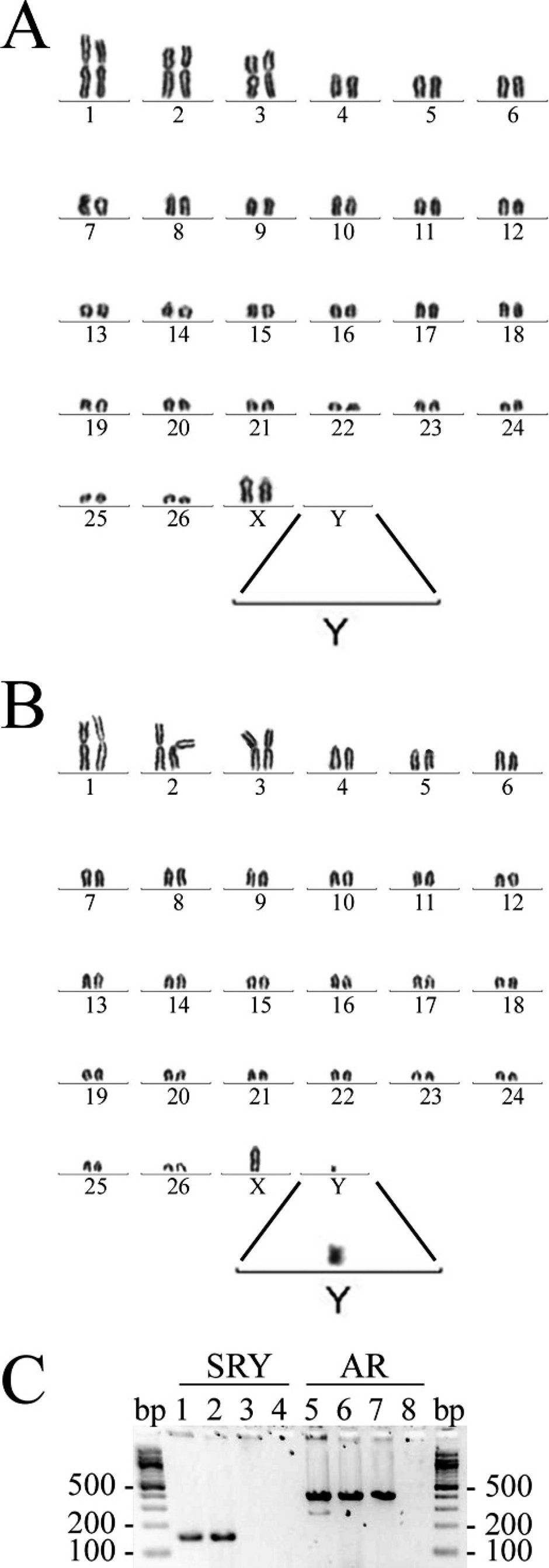
Detection of male twin-derived cells in the peripheral blood of female chimera case 1; Giemsa-stained karyotypes of 2 blood leukocytes.
Sheep scrapie genetic relative susceptibility testing
Samples of blood, ear punch, and hair bulbs obtained from chimera case 1 and from female and male control sheep of various PRNP genotypes were randomly coded and batch submitted to USDA-contract laboratories during the winter of 2007–2008 for sheep scrapie genetic relative susceptibility testing at codon 171. The results for all samples obtained from control sheep were consistent with that derived by PCR sequencing. Results for ear punch and hair bulb samples from chimera case 1 were also consistent with that derived by PCR sequencing. In contrast, 3 different reports were received for testing performed on blood samples from chimera case 1: three laboratories reported this animal's PRNP genotype as RR171, 2 laboratories reported it as QR171, and 1 laboratory (lab no. 6) requested a second blood sample (a duplicate sample from the original blood draw was resubmitted) before reporting that a complex genotype had been detected. After further testing, lab no. 6 reported that 2 genomes were present in the blood sample: one genotyped as QR171 and the other as RR171. Lab no. 6 also requested new hair bulb samples from 3 cases before reporting results; reasons for these requests were not provided.
Chimera cases 2 and 3
Blood samples from these female siblings were subjected to microsatellite analysis, multiplex probing for SRY, and PCR sequencing of PRNP. Reported results from microsatellite analysis were identical for each animal and are presented together in Table 2 (chimera cases 2 and 3). In summary, 1 allele was reported for each of the 3 microsatellites and 2 alleles for each of the 5 microsatellites, but 1 extra allele (3 total) and 2 extra alleles (4 total) were reported for each of the 4 microsatellites and 1 microsatellite, respectively (boldfaced type). “No result” was reported for each of the 2 microsatellites without further explanation. An SRY band was not detected in samples of blood from these 2 sheep nor from a female control sheep but was detected in the blood of a male control sheep; the PRNP band was detected in all blood sample lanes but not the reagent control lane (data not shown). PCR sequencing chromatograms for PRNP codons 136, 154, and 171 were very similar in appearance for blood samples from these 2 sheep (Fig. 3). In each sample, single peaks at each nucleotide position within codons 136 and 154 were consistently observed for both forward (241_SFP) and reverse (12_SRP) sequencing primers; the small nucleotide peaks were not consistently present and represent random background. The corresponding amino acid translations are A136 and R154. In contrast, a polymorphism was consistently detected at codon 171, though again with a minor-A-to-major-G peak appearance at the middle nucleotide position (Fig. 3, green arrows); relevant background peaks were not evident in this vicinity. The polymorphism detected at codon 171 has a corresponding amino acid translation of QR171.
Detection of extra alleles by microsatellite analysis in 4 natural cases of ovine chimerism.*
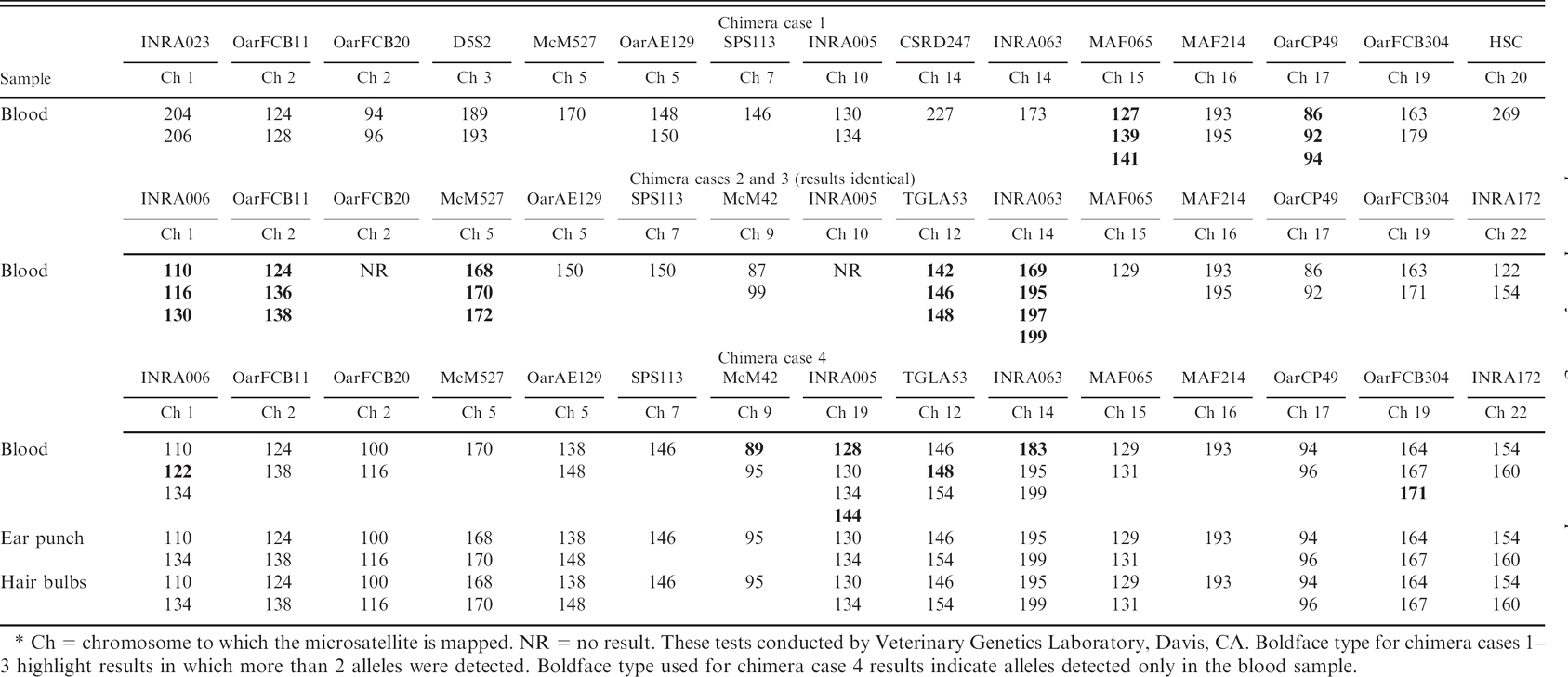
Ch = chromosome to which the microsatellite is mapped. NR = no result. These tests conducted by Veterinary Genetics Laboratory, Davis, CA. Boldface type for chimera cases 1–3 highlight results in which more than 2 alleles were detected. Boldface type used for chimera case 4 results indicate alleles detected only in the blood sample.
Chimera case 4
Blood, ear punch, and hair bulb samples from chimera case 4 were subjected to microsatellite analysis, multiplex probing for SRY, and PCR sequencing of PRNP. Reported results from microsatellite analysis are presented in Table 2 (chimera case 4); for this set of results, extra alleles detected only in the blood sample appear in boldfaced type. Results of microsatellite analyses on samples from the ear punch and hair bulbs were identical, detecting only 1 or 2 alleles for each of the 15 microsatellites; the results for 8 of these microsatellites were identical for the blood sample. An extra (second) McM42 allele, an extra (third) allele for each of the 4 other microsatellites, and 2 extra (third and fourth) INRA005 alleles were also detected in the blood sample. Only a single McM527 allele was detected in the blood sample even though 2 alleles were detected in the ear punch and hair bulb samples.
As shown in Figure 4A, an SRY band was detected in the samples of blood and ear punch from chimera case 4, and from all samples obtained from the male control sheep. An SRY band was not detected in the sample of hair bulbs from chimera case 4 nor was it detected in any of the samples obtained from a female control sheep or in reagent control lanes. PCR sequencing chromatograms for PRNP codons 136, 154, and 171 are shown in Figure 4B. Chromatograms from the ear punch and hair bulb samples showed the same single peaks at each nucleotide position for codons 136, 154, and 171 with corresponding amino acid translations of A136, R154, and R171. The appearance of the chromatogram from the blood at codon 154 was the same as that observed for the ear punch and hair bulb samples. In contrast, additional middle nucleotide peaks were present in the chromatogram from the blood sample at codons 136 and 171 (red and green arrows, respectively; Fig. 4B): minor-thymidine (T)-to-major-cytosine (C) peaks at codon 136 and minor-A-to-major-G peaks at codon 171. This finding was replicated in an additional DNA extraction of the blood sample and was similarly present in all forward and reverse sequencing primer chromatograms (data not shown). Relevant background nucleotide peaks were not evident in the vicinity of these codons. Thus, 2 polymorphisms were detected in the blood sample from chimera case 4 with corresponding amino acid translations of AV136 and QR171.
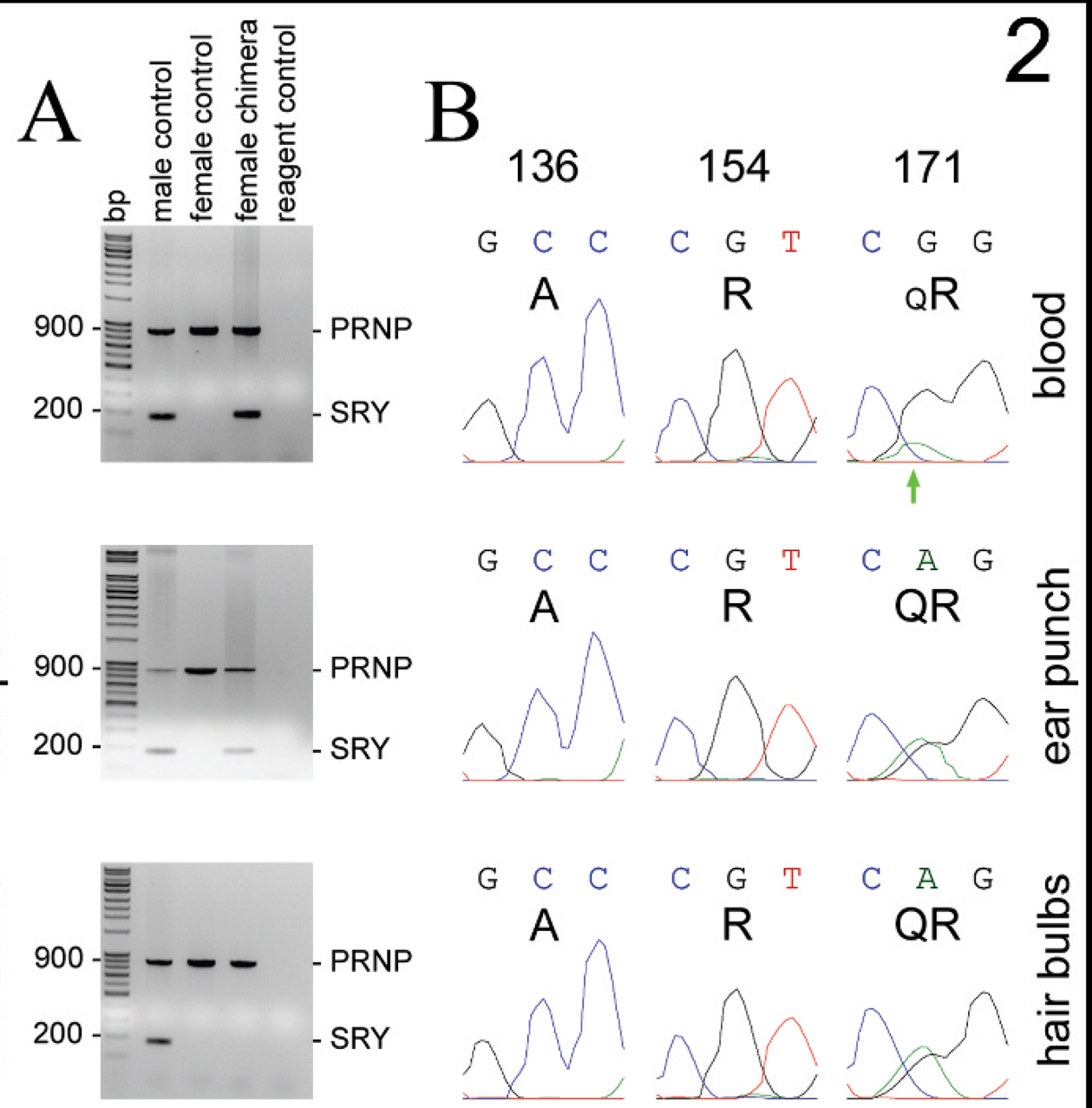
The results of Y-linked SRY detection and corresponding results of PRNP polymorphism detection for chimera case 1. A, in the lane containing DNA extracted from samples of chimera case 1 (female chimera), an SRY band was detected by polymerase chain reaction in the blood and ear punch tissue samples but was not detected in a sample of hair bulbs. SRY was also detected in the male control lane but not in the female control or reagent control lanes. The presence of DNA extract was confirmed by detection of the PRNP band in all lanes except the reagent control lane. Reagent control lanes lack DNA template.
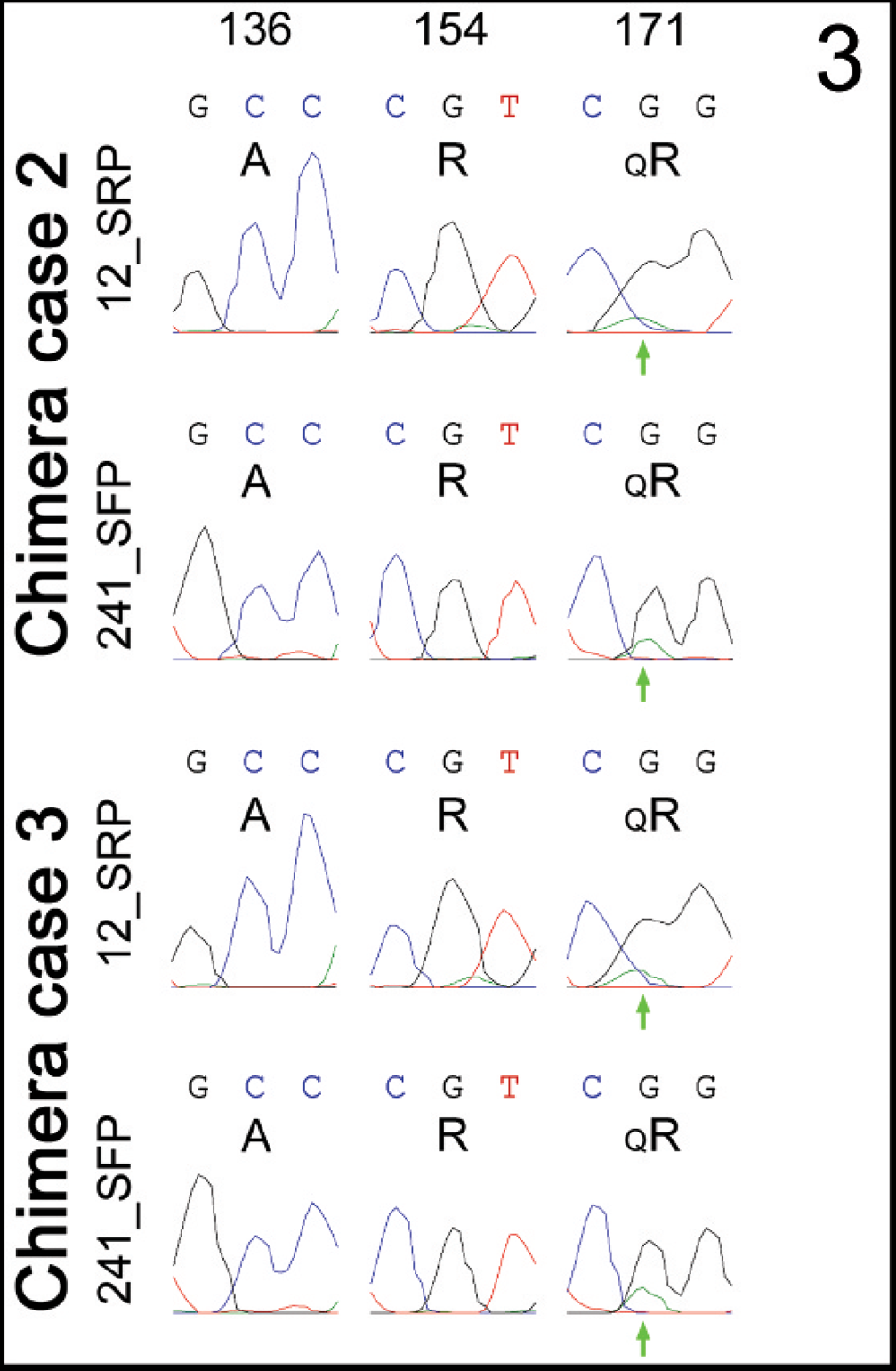
PRNP codon 171 polymorphism as detected in the blood of twin female chimera cases 2 and 3 by polymerase chain reaction sequencing. Note the consistent presence of “imbalanced” nucleotide peaks for the codon 171 polymorphism detected in the forward (12_SRP) and reverse (241_SRP) primer chromatograms of both animals (green arrows). Nucleotide calls and deduced amino acids are shown as described in the legend of Figure 2.
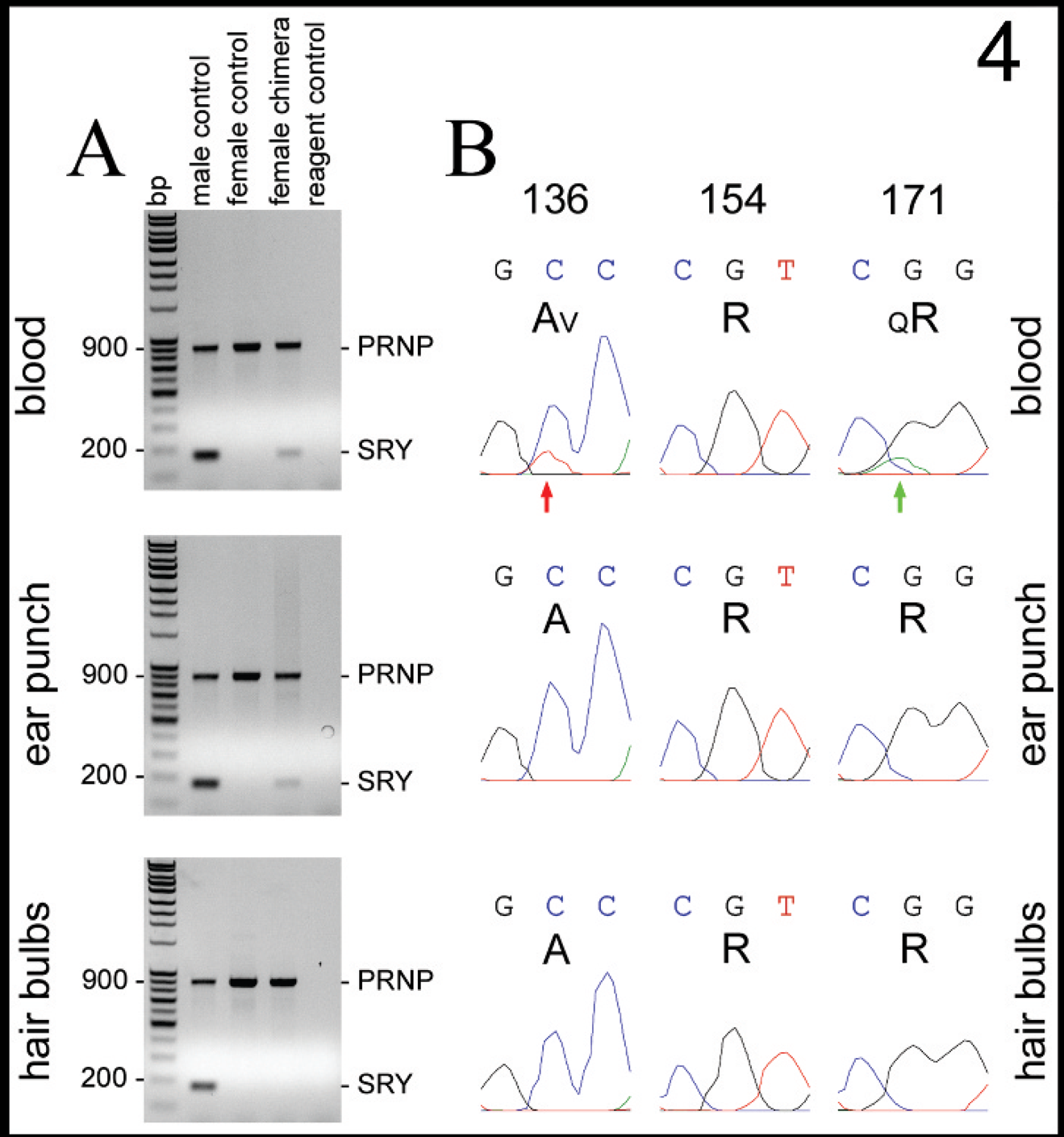
The results of Y-linked SRY detection and corresponding results of PRNP polymorphism detection for chimera case 4.
Discussion
The disposition of ovine chimeras in the U.S. sheep population is of concern to the national scrapie eradication and surveillance programs if such animals confound scrapie disease genetic relative susceptibility testing. In the current report, cytogenetic and molecular analyses were used to identify 4 naturally occurring ovine chimeras in which the PRNP genotypes differed between siblings. The effect of naturally occurring chimerism on PCR sequencing chromatograms for PRNP was similar to the previously described effect of artificial chimerism (ex vivo blood reconstruction) on a genotyping effort utilizing allelespecific probes. 34 Blood samples from chimera case 1 were submitted to 6 contract laboratories routinely conducting scrapie genetic relative susceptibility testing. Three laboratories reported the PRNP genotype of chimera case 1 to be RR171 and 2 reported it to be QR171. Only 1 laboratory reported the result to be “inconclusive” with the later addendum that a complex genotype had been detected after resampling and additional testing. To the authors' knowledge, the present report is the first account to document that naturally occurring ovine chimerism can confound current scrapie genetic relative susceptibility testing.
Similar to reports from the United Kingdom of “complex” PRNP genotypes 13 and “imbalanced” genotyping profiles, 18,26 chimerism in chimera case 1 was originally suspected after routine blood testing by an accredited laboratory. To establish a definitive diagnosis, the authors performed cytogenetic analysis on fresh blood samples obtained from chimera case 1 and demonstrated the presence of 2 populations of diploid leukocytes: one having a normal female chromosome complement and representing the native cell population, the other having a normal male chromosome complement and representing the chimeric cells derived from a dizygotic twin male.
As demonstrated in gender-mismatched female chimera cases 1 and 4, the presence of male twin-derived chimeric cells can also be more simply detected as an amplified Y chromosome linked gene such as SRY. 27 Though the male twins to chimera cases 1 and 4 were not available for study, diagnosis of chimerism in males born twin to females has been similarly achieved through transcript amplification of the X chromosome-linked gene, XIST, 7 a regulatory gene transcribed in XX cells but not in XY cells. 30 From the perspective of chimera detection, however, gender-mismatched chimerism is expected to occur in only about half of all natural cases.
Chimeras can also be detected through commercially available analysis of microsatellites (short tandem repeats), a technique not dependent upon gender mismatch between twins. 27 The microsatellites analyzed in the current study are each present as 2 copies on a single chromosome. Thus, a normal sheep's genome generally carries either 1 or 2 alleles of each of these microsatellites. As illustrated by chimera cases 2 and 3, up to 2 “extra” (or 4 total) alleles may be detected even when the chimeric cells present in the sample are derived from a gender-matched twin. Involvement of multiple microsatellites with extra alleles decreases the chance that a rare gene duplication event has been detected.
As observed in the current study, “imbalanced” polymorphism on PCR sequencing chromatograms can also indicate the presence of chimeric cells. However, the technique has a very limited dynamic range in which to observe allele imbalance and is thus sensitive to the proportion of chimeric cells present in a sample. 26,39 Detection of extra microsatellite alleles is similarly limited. Therefore it should be noted that the percentage of chimeric leukocytes circulating in the blood of sheep can change (decrease or increase) over time and has ranged in female chimeras from 2% to 94%, and in male chimeras from < 1% to 54%. 7,11,19 PCR amplification is a very sensitive technique for detecting low copy number targets in a sample (e.g., SRY in male-derived chimeric cells). 7 As illustrated by female chimera cases 1 and 4, male-derived chimeric cells were detected in ear punch samples by SRY amplification but were not detected by PCR sequencing of PRNP or by microsatellite analysis as applied in the current study. Thus, limited method sensitivity and percentage sample chimerism are factors that may help explain why < 0.1% “complex” genotypes were detected in a national PRNP genotyping effort. 13
Novel technologies are far more sensitive, 39 detecting 10−4% to 10−6% of the minor cell population, but most may not be commercially viable for general application within agriculture. One technology that has the potential for commercial cost-effectiveness might be DNA microarray technology 39 because this platform could be adapted to sensitive detection of unusual genome complexity in addition to trait analyses and other genomics applications. 16,24 None of these methods discriminate natural chimerism from sample contamination between individuals as the cause of detecting a complex genome, but replication of results after controlled resampling diminishes the possibility of inadvertent cross contamination. Because mitochondrial DNA is maternally inherited, 40 codetection of a complex mitochondrial genome might also be used as evidence most consistent with cross contamination. 35
Alternate tissue sampling (e.g., saliva, ear punch, and hair bulbs) is expected to resolve inconclusive results that arise from detection of complex genotypes in blood samples. 18 In cases of natural chimerism, unbiased determination of native cell PRNP genotype optimally requires a tissue sample that lacks chimeric cells. As illustrated in chimera cases 1 and 4, gender-mismatched cells in ear punch samples were able to be detected but not in samples of hair bulbs. This finding should be anticipated since leukocytes are an expected part of the cellular complexity of an ear punch. As such, unbiased determination of native cell PRNP genotype in these 2 chimera cases was only certain by using the samples of hair bulbs. Thus, the native cells of chimera case 1 were determined to be ARQ/ARR, and the native cells of chimera case 4 to be ARR/ARR. Because approximately half of the cells present in the blood of chimera case 1 were derived from the male twin, the genotype of chimeric cells in this female must be ARR/ARR. It was not similarly possible to ascertain the PRNP genotype of chimeric cells in chimera case 4 except that it must include at least 1 VRQ allele.
While determination of native cell genotype in natural chimeras may serve some limited purposes, effective detection of chimerism would seem the preferred goal for national scrapie eradication efforts as well as for broader application of molecular genetics to other livestock selection strategies. 16,24 Where detection of chimeras is deemed an appropriate adjunct to other genetic testing being performed, the results of the current study suggest blood samples are preferable.
With the goal of eradicating scrapie disease, genetic relative susceptibility testing in sheep helps guide the rational selection of resistant stock and removal of susceptible stock. Naturally occurring chimeras confound such testing. Estimates of chimera prevalence among progeny of multiparous births have ranged from 3% to 5% in some common breeds of sheep, but up to 25% in certain high-fecundity breeds. 7,10,17,19,22,23,25,37,38 Considering that chimerism is unexpected in singlet lambs, and utilizing 2001 lambing outcomes for females across the United States (USDA: 2003, Part III: lambing practices, spring 2001. Available at http://www.aphis.usda.gov/vs/ceah/ncahs/nahms/sheep/sheep01/sheep01Pt3.pdf. Accessed on January 20, 2009), the average prevalence of chimerism can be roughly estimated to be 2–4% the annual lamb crop.
Undetected chimeras undermine efforts to select genetically resistant replacement stock. Even though chimeric females born twin to males are most often infertile, 28 female chimeras born twin to other dizygotic females, and male chimeras born twin to either gender, are generally fertile. 21,22 In the present study, the twin females (chimera cases 2 and 3) and the males born twin to chimera cases 1 and 4 were all likely to be fertile. Considering the confounding effect of chimeras on genetic relative susceptibility testing, it is conceivable that fertile male and female chimeras may be sometimes reported as the highly resistant PRNP genotype, ARR/ARR, and yet contribute a susceptible haplotype (ARQ or VRQ) to their progeny. This is most likely to occur when chimerism goes undetected and the genotype of chimeric cells is reported, such as was reported by 3 of the 6 laboratories for chimera case 1 in the present study. Reports of chimeric gamete-forming cells suggest unexpected progeny could be produced by undetected chimeras whose native cell PRNP genotype is reported. 7,28,33
Undetected chimeras undermine efforts to properly identify and remove susceptible scrapie-exposed animals. The genetic relative susceptibility of a chimera whose native and chimeric cell populations have the same PRNP genotype might be reasonably assumed to be the same as a normal sheep of that genotype. However, the genetic relative susceptibility of a chimera whose native and chimeric cell populations differ in PRNP genotype cannot be assumed to be the same as a normal heterozygote sheep. For example, the scrapie susceptibility of an ovine chimera whose complex genome includes cells with the resistant genotype ARR/ARR and others with the susceptible genotype ARQ/ARQ cannot be safely estimated to be equal with the relatively high degree of scrapie disease resistance in ARQ/ARR sheep. 12 In experiments that might mimic natural tissue chimerism, prions have been shown to replicate within genetically susceptible cells engrafted in tissues otherwise composed of scrapie-resistant cells. 1,2,8 Application of methodology to detect genetically susceptible cells in an environment of resistant cells could help determine if natural tissue chimerism might similarly support prion replication in sheep.
In summary, the current study documents the confounding effects of natural ovine chimerism on scrapie genetic relative susceptibility testing, highlighting the potential importance of chimera detection to current national scrapie eradication efforts both in the United States and abroad. While proper identification of all ovine chimeras may not be necessary to achieve the goal of scrapie disease eradication in the United States, efforts to detect chimeras may be warranted in certain situations. For example, the increased rate of chimerism in sheep of certain breeds or under certain modern husbandry management practices 29 may warrant special evaluation for chimerism. Special evaluation may be warranted as well where selection of genetically resistant breeder stock is highly valued. Until proven otherwise, it seems prudent to consider chimeras that harbor cells with a scrapie-susceptible PRNP genotype to be at risk for contracting scrapie disease, and to consider special evaluation to detect chimeras in scrapie-exposed flocks. It is likely that the confounding effects of chimerism will be relevant to other species in which natural chimerism has been observed, such as goats, cervids, and cattle. 28 Thus, detection of chimeras will be similarly relevant to any future application of genetic relative susceptibility testing to transmissible spongiform encephalopathies in these species, but also to any cost-effective development of a more broadly applied molecular genetics platform.
Acknowledgements
This work was supported by CRIS 5348–32000-021-00D from the Agricultural Research Service, USDA. The authors especially wish to recognize the logistic and expert technical contributions of Desiree Lesiak to this project, and thank Caylee Birge, Duane Chandler, Amy Hetrick, and Marta Henrickson for their contributions to the care and use of the animals. From the USDA-Animal and Plant Health Inspection Services, the authors thank Dr. John V. Duncan (Veterinary Services), and Norma J. Newton and Patricia A. Meinhardt (both from the Pathobiology Laboratory, National Veterinary Services Laboratory), for their interest and support in helping to obtain chimeric animals or samples for study. The authors thank Patricia Meinhardt, Dr. Diane Sutton (National Scrapie Program Coordinator, National Center for Animal Health Programs), and Associate Professor Mike Varnum (Washington State University) for their critical review of the manuscript. Mention of trade names or commercial products or enterprises in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA.
Footnotes
a.
USDA-Animal and Plant Health Inspection Service-Veterinary Services-National Veterinary Services Laboratories, Ames, IA.
b.
USDA-Agricultural Research Service-Pacific West Area-Animal Disease Research Unit, Pullman, WA.
c.
Vacutainer® System, BD, Franklin Lakes, NJ.
d.
Typifix sample collection system, Agrobiogen GmbH, Hilgertshausen, Germany.
e.
Molecular Cytogenetics Laboratory, Texas A&M University, College Station, TX.
f.
Invitrogen Corp., Carlsbad, CA.
g.
Gemini Bio-Products, West Sacramento, CA.
h.
Sigma-Aldrich, St. Louis, MO.
i.
Rainbow Scientific Inc., Windsor, CT.
j.
Axioplan 2, Carl Zeiss MicroImaging GmbH, Göttingen, Germany.
k.
Ikaros Karyotyping System (version 5.2.2), MetaSystems GmbH, Altlussheim, Germany.
l.
JumpStart™ REDTaq®, Sigma-Aldrich, St. Louis, MO.
m.
Gentra® Puregene® DNA extraction kit, Qiagen Inc., Valencia, CA.
n.
Taq PCR Core Kit, Qiagen Inc., Valencia, CA.
o.
ExoSAP-IT®, USB Corp., Cleveland, OH.
p.
Laboratory for Biotechnology and Bioanalysis, Washington State University, Pullman, WA.
q.
SeqMan Pro, DNASTAR Inc., Madison, WI.
r.
Veterinary Genetics Laboratory, University of California, Davis, CA.