
Abstract
Although the widespread occurrence of porcine group C rotaviruses (GCRV) is assumed, precise prevalence remains largely unknown because of the absence of reliable, specific, and rapid diagnostic methods. To detect and quantify porcine GCRV, the authors evaluated and optimized SYBR Green and TaqMan real-time reverse transcription polymerase chain reaction (RT-PCR) assays and applied them to 108 piglet fecal samples. Using serially diluted standard RNA transcripts of porcine GCRV VP6 gene, both SYBR Green and TaqMan real-time RT-PCR assays detected as few as 1 × 101 genome copies/μl (correlation coefficiency >0.99), whereas conventional RT-PCR detected 1.0 × 103 copies/μl. In addition, the conventional assay detected porcine GCRV in 24% (26/108) of fecal samples, whereas the detection rates of both SYBR Green and TaqMan assays were 72% (78 of 108) and 64% (70 of 108), respectively. The current study indicated that both real-time RT-PCR assays were reliable, specific, and rapid methods for the detection of porcine GCRV in porcine fecal samples.
Keywords
Rotaviruses are an important cause of acute gastroenteritis transmitted by the oral-fecal route. 5 Rotaviruses are members of the Reoviridae family and are classified into at least 7 groups (A–G) by multiple serologic methods. 5 The genome consists of 11 segments of double-stranded RNA that encode 6 structural and 6 nonstructural proteins. 5 Serogroups A, B, and C are known to cause gastrointestinal infections in humans, but most human infections belong to group A. 5 Group B rotaviruses cause adult diarrhea and are geographically confined. 9 Group C rotaviruses (GCRVs) were first recognized as a causative agent of gastroenteritis in pigs in the 1980s 3 and were subsequently identified as an emerging human pathogen. 11,14,16,17 Epidemiologic studies suggest that porcine GCRVs are widespread in pig herds and are presumably zoonotic. 18 However, the precise prevalence of porcine GCRVs remains largely unknown because of the absence of reliable, specific, and rapid diagnostic methods.
Various methods are applied to detect rotaviruses in feces, which may have a high virus level during the acute phase of the disease. Negatively stained virions can be detected by electron microscopy, 2 and virus antigen can be detected by enzyme immunoassay, 1 passive particle agglutination tests, 15 and lateral flow immunoassays. 19 Direct detection of the viral genome can be done by reverse transcription polymerase chain reaction (RT-PCR). 8 However, these routine methods are also limited. For example, electron microscopy does not define serotype and is time-consuming and complex. 6 In contrast, enzyme immunoassay and conventional RT-PCR are relatively fast, sensitive, and specific but not appropriate for samples with a low concentration of the virus. 12 Both techniques are only qualitative, while the latter requires procedures such as Southern hybridization with specific probe for confirmation. 6
Real-time PCR has several advantages over conventional PCR for measuring the amount of pathogen nucleic acid in diagnostic samples. These include highly a reduced possibility of contamination due to carryover, reduced risk of exposure to ethidium bromide, and a less cumbersome procedure requiring no gel electrophoresis. 6 In the current study, SYBR Green and TaqMan real-time RT-PCR assays were developed for the rapid, specific, and highly sensitive detection of porcine GCRVs from the stool samples of pigs.
One hundred eight diarrheic fecal samples were obtained from weaning and postweaning pigs in South Korea during 2006 to 2008. 10 For the detection of porcine GCRV, 0.1 g of feces was suspended in 0.9 ml phosphate buffered saline, yielding a 10% suspension. 10 The mixture was vortexed for 1 min and centrifuged at 2,060 × g for 10 min at 4°C. The viral RNA was purified from 200 μl of fecal mixture using a viral RNA mini kit a according to the manufacturer's instructions. To reduce the activity of PCR inhibitors, the extracted RNA was diluted 10 times with diethylpyrocarbonate-treated water. The viral RNA was stored at −80°C until further analysis.
List of primers and probe used for conventional real-time reverse transcription polymerase chain reaction (RT-PCR) and SYBR Green and TaqMan real-time RT-PCR assays for the detection and quantification of porcine group C rotaviruses. *

F = forward primer; R = reverse primer; Pr = probe.
The 1-step RT-PCR was performed with the oligonucleotide primer pair 7 specific to a 356-bp fragment of GCRV VP6 gene (Table 1) as described previously. 10 The SYBR Green and TaqMan real-time RT-PCR assays were performed with primers and probe (Table 1), which were designed from the published sequence of the highly conserved region of porcine GCRV VP6 gene. b Both realtime RT-PCR assays were carried out using a commercial real-time amplification system c and a commercial 1-step RT-PCR kit. d Briefly, 5 μl of extracted RNA were added to 25 μl of master mix with primers at 300 nM final concentration for the SYBR Green real-time RT-PCR assay. For the TaqMan assay, probe (150 nM final concentration) was added into the primer containing the master mix. The SYBR Green real-time RT-PCR conditions were 30 min at 42°C, 10 min at 95°C, 45 cycles of denaturation at 95°C for 15 sec, annealing at 55°C for 30 sec, and extension at 72°C for 20 sec. The conditions for the TaqMan real-time RT-PCR assay were 30 min at 42°C, 10 min at 95°C, 45 cycles of 10 sec at 95°C, 15 sec at 50°C, 30 sec at 57°C, and 5 sec at 72°C. In this TaqMan PCR cycle, 2 annealing temperatures (15 sec at 50°C and 30 sec at 57°C, respectively) were employed to augment the detection rate. Quantitative detection of porcine GCRVs was performed as separate reactions in duplicate.
To evaluate and optimize the real-time RT-PCR assays, complementary RNA standards of porcine GCRV were generated using the amplified RT-PCR products of a 356-bp fragment of porcine GCRV VP6 gene from porcine GCRV-positive fecal samples. After gel elution of the RT-PCR product, the amplicon was cloned into a cloning vector e and propagated in Escherichia coli. Plasmid DNA was purified using a mini prep kit. f After the plasmid DNA was digested with Pst I to generate a blunt end, DNA was purified using a PCR purification kit. f The linearized DNA clone was used as the template for in vitro transcription using the MEGAscript kit g according to the manufacturer's instructions. Synthesized complementary RNA (cRNA) was stored at −80°C until required. Standard curves were generated using 10-fold serial dilutions from 101 to 108 copies of cRNA. Experiments were performed in duplicate.
All statistical analyses were carried out with SPSS version 11.5.1 for Windows. h The limit of statistical significance for the conducted tests was defined as P < 0.05. The kappa coefficient was calculated to assess the agreement between real-time RT-PCR assays and conventional RT-PCR assay.
In a BLAST search (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi), the primers and probe used in the SYBR Green and TaqMan real-time RT-PCR assays did not reveal any sequence similarities besides GCRV (data not shown). To confirm the specificity of the primers and probe, both realtime RT-PCR assays were also performed with RNA extracted from cell lines infected with Transmissible gastroenteritis virus, Porcine epidemic diarrhea virus, Porcine group A rotavirus, and Porcine sapovirus; with DNA extracted from Escherichia coli and Salmonella typhimurium isolated from diarrheic fecal samples; and with fecal samples containing Lawsonia intracellularis or Brachyspira hyodysenteriae. Neither real-time RT-PCR assays detected cross-amplification in these control samples (data not shown), indicating that these assays were specific to porcine GCRV.
To determine the analytical sensitivity and specificity, in vitro RNA transcripts of porcine GCRV were generated using serial dilution of the standard RNA transcripts from 1.2 × 101 to 1.2 × 108 copies/μl. The slope values of the standard curves with SYBR Green and TaqMan were −3.34 and −3.73, respectively. The melting point of the SYBR Green assay was 79°C (SD: 0.13°C). Conventional RT-PCR detected standard RNA copies to 1 × 103 copies/μl of standard RNA. Therefore, both real-time RT-PCR assays were 100 times more sensitive, detecting standard RNA copies to 1 × 101 copies/μl. Only the expected band was observed on agarose gel electrophoresis.
All 3 assays, conventional RT-PCR, SYBR Green, and TaqMan RT-PCR, were used to detect porcine GCRV in 108 diarrheic fecal samples (Table 2). Conventional RT-PCR detected porcine GCRV in 26 (24%) of 108 stool samples, whereas SYBR Green and TaqMan real-time RT-PCR assays detected porcine GCRV in 78 (72%) and 70 (64.8%) of 108 stool samples, respectively. The samples determined to be positive by the conventional PCR were also positive by the 2 real-time RT-PCR assays. All amplicons from positive PCR reactions on both real-time RT-PCR assays were directly sequenced and found to match the nucleotide sequence of porcine GCRV VP6 gene (data not shown), confirming that these were specific reactions. Given the results from the limit of detection assay and field diarrheic samples, both the SYBR Green and TaqMan RT-PCR assays are reliable, reproducible, and sensitive tools for the rapid detection of porcine GCRVs in swine stool samples.
Comparison of the detection rate of porcine group C rotaviruses by SYBR Green and TaqMan real-time reverse transcription polymerase chain reaction (RT-PCR) assays with the conventional RT-PCR assay.
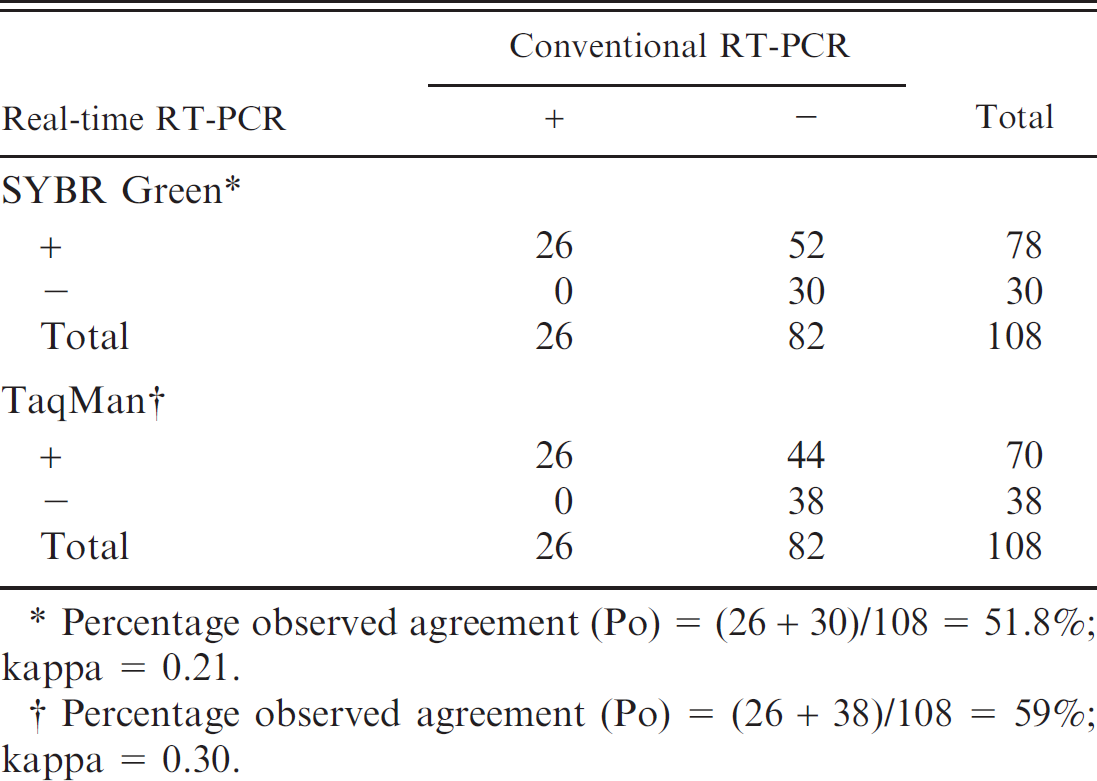
Percentage observed agreement (Po) = (26 + 30)/108 = 51.8%; kappa = 0.21.
Percentage observed agreement (Po) = (26 + 38)/108 = 59%; kappa = 0.30.
The authors previously reported that the prevalence of porcine GCRV infections in South Korea is 26% of the surveyed diarrheic fecal samples by conventional RT-PCR. 10 However, the present findings by both real-time RT-PCR assays indicate that the fecal prevalence of the porcine GCRVs in piglet diarrhea is much higher than that reported previously in South Korea. 10
Although RT-PCR has been used for the detection of porcine GCRV, 4,10 this assay cannot measure the viral load in the stool samples, resulting in a significant gap in veterinary knowledge about virological and epidemiological features. Therefore, the copy number of porcine GCRV in the stool samples by real-time RT-PCR assays was evaluated. From all positive samples, both SYBR Green and TaqMan real-time RT-PCR assays detected porcine GCRV RNA with values of mean threshold cycle (Ct) ranging from 7 to 37 and 8 to 37 Ct, respectively. Forty-four fecal samples that tested positive by both real-time RT-PCR assays but negative by conventional RT-PCR assay contained lower numbers of porcine GCRV RNA (range: 2.1 × 102 to 3.8 × 103 copies/μl) than those testing positive by RT-PCR assay.
In contrast to the standard melting point curve of the SYBR Green real-time RT-PCR assay with in vitro cRNA transcripts, greater variations were evident with the field samples, ranging from 74.5°C to 80°C (data not shown). Factors attributed to this dichotomy include initial concentration, sequence and size of the template, GC content, and presence of interference materials in fecal samples. 13
In this study, the SYBR Green assay was found to be more sensitive than the TaqMan assay (Table 2). Porcine GCRVs are thought to mutate at a high frequency, resulting in high error rates of the RNA polymerase similar to other RNA viruses. 5 This diversity in gene segments makes it difficult to detect all strains using a universal probe. To reduce false-negative results related to strain variation, TaqMan oligonucleotides should be designed in a highly conserved region of the VP6 gene or a degenerate probe should be used.
In conclusion, the current study reports the development of rapid, sensitive, specific, and reproducible 1-step SYBR Green and TaqMan real-time RT-PCR assays for detection of GCRVs in porcine fecal samples. Both assays were found to be more sensitive than conventional RT-PCR and would be useful tools for diagnosing GCRV infections in farms with enteritis in weaning and postweaning piglets.
Acknowledgements. This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant (2009-0081752) and the Regional Technology Innovation Program (RTI05-01-01) of the Ministry of Knowledge Economy (MKE), Republic of Korea. The authors acknowledge a graduate fellowship provided by the Korean Ministry of Education and Human Resources Development through the Brain Korea 21 project.
Footnotes
a.
Bioneer, Daejon, South Korea.
b.
GenBank accession no. M94157.
c.
RG-6000 Rotor-Gene Real Time Amplification system, Corbett Research, Mortlake, Australia.
d.
SensiMix one-step RT-PCR kit, Quantace Ltd., London, UK.
e.
yT&A®, Yeastern Biotech Co. Ltd., Taipei, Taiwan.
f.
Qiagen Inc., Valencia, CA.
g.
Ambion, Austin, TX.
h.
SPSS Inc., Chicago, IL.