
Abstract
Enterococcus cecorum is an emerging challenge to the broiler industry. The organism has been implicated in septicemia, spondylitis, arthritis, and osteomyelitis in commercial broilers and broiler breeders, which lead to economic losses attributed to increased mortality and culling rates, decreased average processing weights, and increased feed conversion ratios. The current study evaluated the genetic variability of 30 clinical isolates of E. cecorum from outbreaks in Pennsylvania, using 3 molecular typing methods, namely, pulsed-field gel electrophoresis (PFGE), randomly amplified polymorphic DNA analysis, and enterobacterial repetitive intergenic consensus–PCR (polymerase chain reaction), in order to understand their genetic relatedness and to identify possible pathogenic clones. The study revealed the existence of genotypic polymorphism among E. cecorum associated with clinical disease. Of the 3 typing methods used, PFGE analysis demonstrated higher genetic variability of E. cecorum isolates compared to PCR-based methods. Also, each molecular typing method was evaluated in terms of typeability, discriminatory power, and reproducibility for application of these typing methods in fingerprinting of E. cecorum in future reference. Pulsed-field gel electrophoresis provided the most reliable results with greater discriminatory power and higher reproducibility compared to the 2 PCR-based methods.
Introduction
Enterococcus cecorum, which was formerly known as Streptococcus cecorum, is an emerging pathogen of the broiler industry. The organism was first isolated from chicken intestinal flora in 1983. 4 Enterococcus cecorum is a facultatively anaerobic, catalase-negative, alpha-hemolytic, Gram-positive coccus that belongs to an atypical group of enterococci that do not grow in 6.5% NaCl. 3 Even though E. cecorum was previously known as a commensal in poultry, it has recently been recognized as an important pathogen of commercial broilers and broiler breeders. Infection with E. cecorum is apparent in various forms where spondylitis, osteomyelitis, arthritis, and septicemia are the most predominant signs.12,13 Sporadic cases of septicemia, endocarditis, and osteomyelitis caused by E. cecorum have also been reported in immune-compromised human patients.1,7,14
Despite the acknowledged status of the organism as a poultry pathogen, the exact mode of transmission, port of entry, and virulence mechanisms of E. cecorum are largely unknown. Therefore, appropriate epidemiological surveillance is a necessity for disease investigation, and implication of preventive and control measures in an outbreak situation. Genotyping to determine the clonal diversity or to track a particular clonal type plays a pivotal role in epidemiological surveillance. Although various genomic “fingerprinting” approaches have been widely used for these purposes with other bacteria, there is no typing method available for E. cecorum. Accordingly, the objective of the current study was to analyze the genetic variability of E. cecorum and to evaluate its genetic relatedness in relation to the source of infection.
Various nucleic acid–based techniques have been used to type and characterize the genetic variability of enterococci.5,6,16 Such molecular techniques include pulsed-field gel electrophoresis (PFGE), plasmid profiling, and polymerase chain reaction (PCR)-based methods such as randomly amplified polymorphic DNA PCR (RAPD-PCR), repetitive extragenic palindromic sequence PCR (Rep-PCR), and enterobacterial repetitive intergenic consensus PCR (ERIC-PCR). Of these methods, PFGE is considered the “gold standard” for molecular typing of many bacterial species. 15 The PFGE SmaI microrestriction patterns of enterococci have been used extensively for genotyping and fingerprinting of other species of enterococci.9,15,16 Similarly, RAPD-PCR, Rep-PCR, and ERIC-PCR also have been used for molecular characterization of enterococci species present in dairy products and sea foods, as well as in clinical settings.2,9,16 In the present study, 3 typing methods, namely, PFGE, RAPD-PCR, and ERIC-PCR, were used for the purpose of genotyping and assessing the genetic relatedness of 30 isolates of E. cecorum associated with clinical disease.
Materials and methods
Isolation and identification of E. cecorum strains
Bacteria were isolated from clinical cases submitted to the Animal Diagnostic Laboratory at the Pennsylvania State University (University Park, Pennsylvania) from 2008 to 2011. For bacterial isolation, tissue samples were inoculated onto Columbia colistin and nalidixic acid agar plates with 5% sheep blood and incubated at 37°C for 48 hr. Bacteria were presumptively identified as E. cecorum by Gram-staining, an automated identification system, a growth characteristics in 6.5% NaCl broth, b and ability to ferment raffinose. c Presumptively identified bacteria were confirmed as E. cecorum by 16S ribosomal RNA gene sequencing. 8
PFGE procedure
Preparation of DNA plugs of E. cecorum was performed with a previously described rapid DNA preparation procedure with minor modifications. 10 Briefly, bacterial pellets of 2.5 ml of overnight cultures were resuspended in 0.5 ml of 2× lysis solution containing 12 mM of Tris-HCl (pH 7.4), 2 M of NaCl, d 20 mM of ethylenediamine tetra-acetic acid (EDTA; pH 7.5), 1% Brij, e 0.4% deoxycholate, e 1% sodium lauroylsarcosine, f 1.0 mg/ml of lysozyme, d and 100 µg/ml of RNase A. g Immediately after resuspension of the bacterial pellet, 0.5 ml of 1.6% pulsed field certified agarose, h cooled down to 50°C, was added. The mixture was vortexed briefly before being loaded into the plug molds. h Once the plugs were solidified, the plugs were lysed first in 3 ml of 1× lysis solution containing 0.5 mg/ml of lysozyme d and 100 µg/ml of RNase g at 37°C for 2 hr with gentle shaking (100 rpm/min) and then in 3 ml of solution containing 10 mM of Tris (pH 7.4), 1 mM of EDTA, 100 mg/ml of proteinase K, f and 1% sodium dodecyl sulfate d at 50°C for 1 hr. Finally, the plugs were washed twice with 7 ml of TE buffer (10 mM Tris-HCl [pH 7.4] and 0.1 mM of EDTA) at 50°C for 30 min and stored in 5 ml of TE buffer until used. According to the literature, enterococci genomes have a low G+C content 11 ; therefore, restriction digestion was performed using SmaI g for 2 hr at 25°C, and bands were separated using a commercial PFGE system h on 1.5 % pulsed-field certified agarose h for 24 hr at 14°C with switching times ramped from 0.5 to 35 sec and a gradient of 6 v/cm. Enterococcus faecalis American Type Culture Collection (ATCC) 29212 was used as the molecular weight size standards. The restriction patterns were visualized by ultraviolet transillumination. i
RAPD-PCR and ERIC-PCR procedure
Extraction of genomic DNA was performed using a genomic DNA extraction kit g according to the manufacturer’s instructions. Both RAPD-PCR and ERIC-PCR were performed as previously described. 9 The RAPD-PCR assay was carried out in 50-μl reaction volumes containing 3 mM of MgCl2, 400 μM of deoxyribonucleotide triphosphates (dNTPs), 1 μM of M13 primer (5′-GAGGGTGGCGGTTCT-3′), 1.25 U of Taq DNA polymerase, j and 200 ng of template DNA in 1× PCR buffer. j Cycling parameters used for amplification were an initial denaturing at 94°C for 60 sec, followed by 35 cycles of 94°C for 60 sec, 40°C for 20 sec, and 72°C for 80 sec, and final extension at 72°C for 5 min. The ERIC-PCR reactions were carried out in 25-μl reaction volumes containing 3 mM of MgCl2, 400 μM of dNTPs, 1 μM of ERIC1-R primer (5′-ATGTAAGCTCCTGGGGATTCAC-3′), 1.25 U of Taq DNA polymerase, j and 200 ng of template DNA in 1× PCR buffer. Amplification conditions were as follows: initial denaturing at 94°C for 3 min, 35 cycles of 94°C for 30 sec, 48°C for 60 sec, and 72°C for 5 min, and final extension at 72°C for 7 min. The PCR products were analyzed following electrophoresis on 1.5% agarose k at 120 V for 2 hr in 1× TAE buffer (40 mM of Tris-acetate, 2.5 mM of EDTA; pH 8). Gels were stained with ethidium bromide, d and the images were captured using a gel documentation system. i
Data analysis
Banding patterns obtained in each typing method were analyzed using a commercial software. l Similarity percentages of fingerprints were analyzed using dendrograms constructed by Dice coefficient and the unweighted pair group method with arithmetic averages with 1.5% band position tolerance. Similarity index >80% was used to assign genotypes with each method. Simpson Index was used to evaluate the discriminatory power of each molecular typing method (http://darwin.phyloviz.net/ComparingPartitions/index.php).
Results
Enterococcus cecorum strains
A total of 27 E. cecorum strains isolated from commercial broilers and broiler breeders with osteomyelitis (n = 6), spondylitis (n = 12), septicemia (n = 2), arthritis (n = 5), cellulitis (n = 1), and yolk sac infection (n = 1) were used in the current study (Table 1). An additional 3 strains were isolated from cecal contents of a commercial broiler with spinal abscesses. Origin and source of the isolates are summarized in Table 1.
The source, origin, and genotype of Enterococcus cecorum isolates submitted to the Animal Diagnosis Laboratory, The Pennsylvania State University (University Park, PA) during 2008 to 2011.*
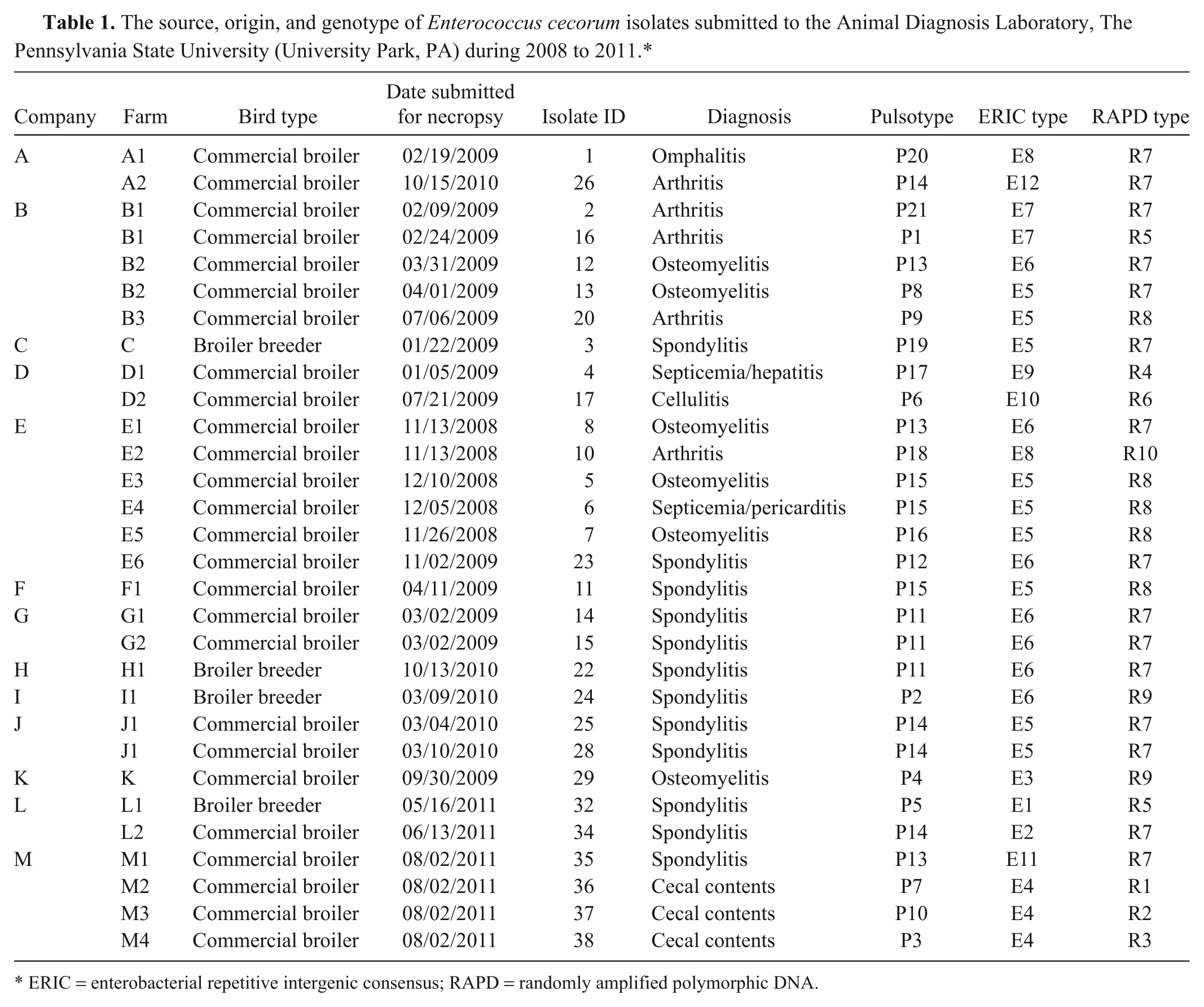
ERIC = enterobacterial repetitive intergenic consensus; RAPD = randomly amplified polymorphic DNA.
PFGE analysis
SmaI restriction digestion of E. cecorum generated 12–16 fragments with molecular weights ranging approximately from 10 kb to 400 kb (Fig. 1A). Based on >80% similarity index, 21 pulsotypes were observed for the 30 E. cecorum isolates included in the study (Table 1, Fig. 1B). Within the 21 pulsotypes, 17 isolates had unique fingerprints. A total of 4 isolates showed the fingerprints of pulsotype 14, and pulsotypes 11, 13 and 15 enclosed 3 isolates within each group with similarity ranging from 85% to 100%. The discriminatory power of PFGE was high with a Simpson Index of 0.966 (95% confidence interval [CI]: 0.937–0.994). Upon repeated testing, PFGE provided exactly the same banding patterns ensuring the reproducibility of the test (see supplemental data).
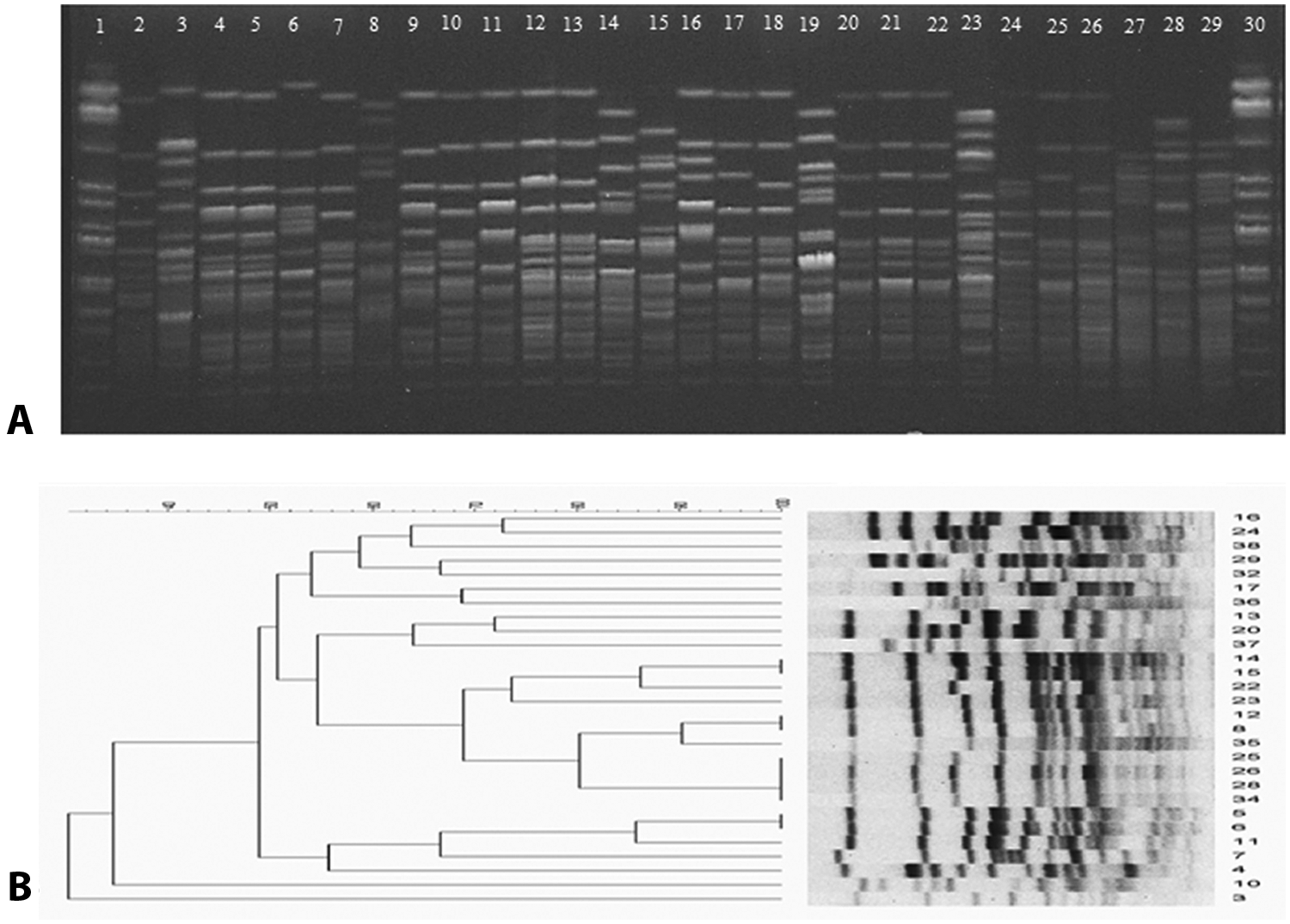
Results of pulsed field gel electrophoresis (PFGE) examination of Enterococcus cecorum clinical isolates.
ERIC-PCR analysis
The ERIC-1R primer generated 2–7 amplicons with molecular weights ranging from 200 to 5,000 bp (Fig. 2A). According to the dendrogram with >80% similarity, 12 different genotypes were observed (Table 1, Fig. 2B). Of the 30 isolates tested, 7 isolates provided unique genotypes (E1, E2, E3, E9, E10, E11, and E12) whereas genotype E5 contained the highest number of isolates (n = 9) with similarity ranging from 81% to 100%. The Simpson Index of the ERIC-PCR was 0.857 (95% CI: 0.770–0.938). Based on the 95% CI, statistically no difference was observed in the discriminatory power between ERIC-PCR and PFGE. However, the reproducibility that ERIC-PCR demonstrated was inferior to that of PFGE (see supplemental data).
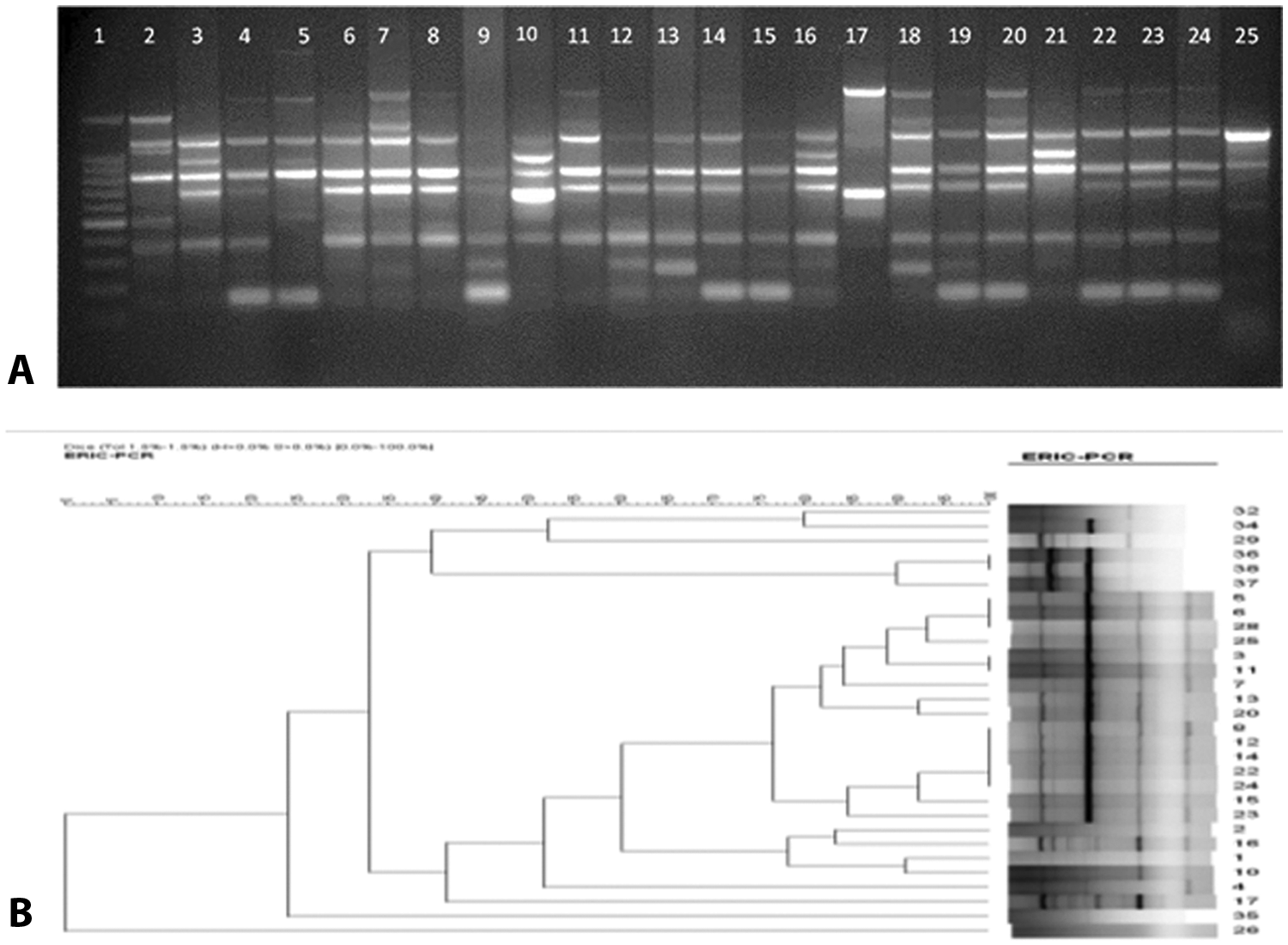
Results of enterobacterial repetitive intergenic consensus–polymerase chain reaction (ERIC-PCR) analysis of Enterococcus cecorum clinical isolates.
RAPD-PCR analysis
The M13 primer yielded amplicons ranging from 750 bp to 5 kb (Fig. 3A). Ten unique genotypes were observed for the 30 E. cecorum isolates tested (Table 1). The majority of isolates (46.66%) belonged to the RAPD genotype 7, with a similarity ranging from 85% to 100% (Fig. 3B). The RAPD-PCR exhibited the lowest discriminatory power, which was 0.731 (95% CI: 0.577–0.885) and the lowest reproducibility compared to both PFGE and ERIC-PCR methods (see supplemental data).
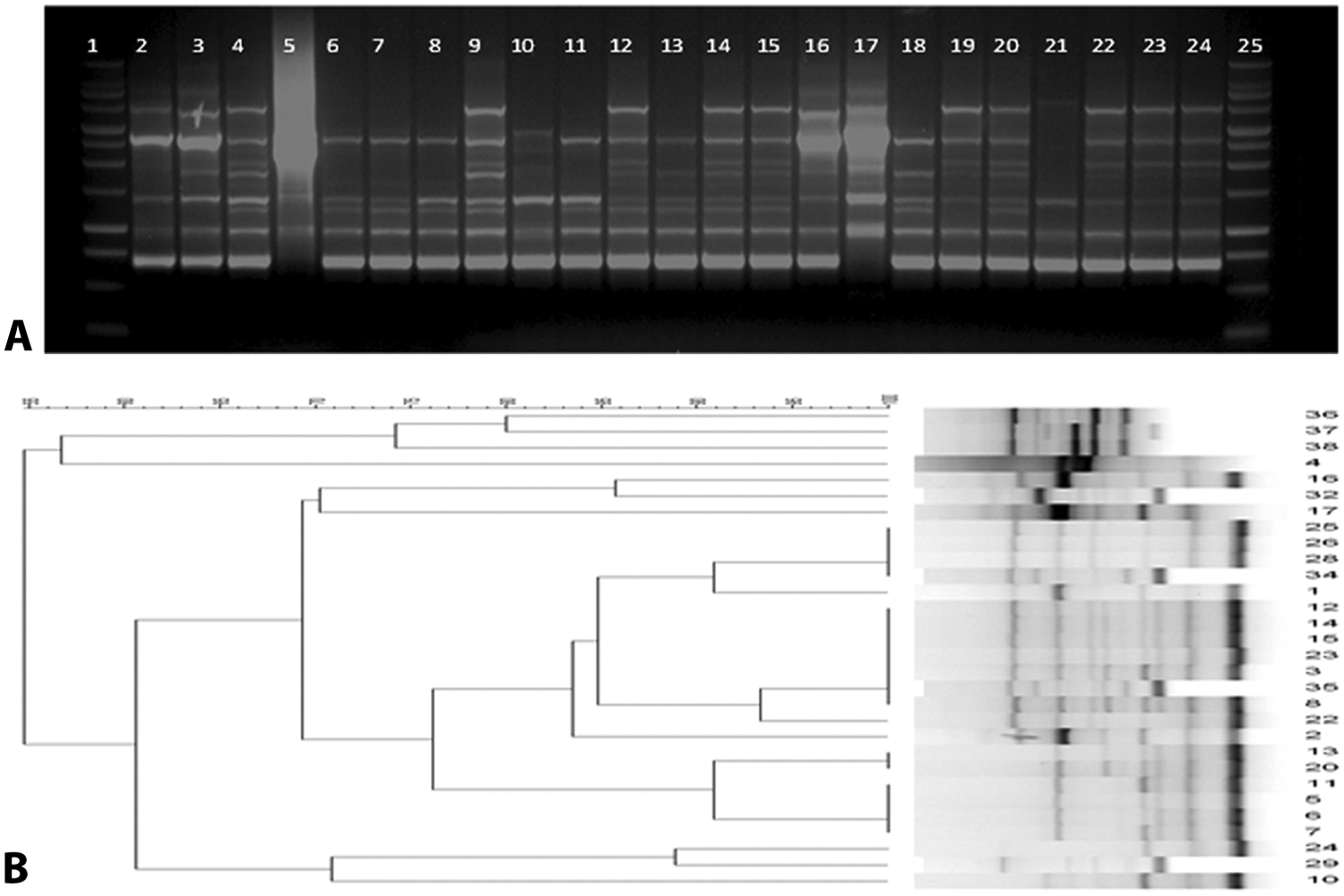
Results of randomly amplified polymorphic DNA (RAPD) analysis of Enterococcus cecorum clinical isolates.
Discussion
Enterococcus cecorum is an emerging pathogen of concern to the broiler industry. Previously, it was recognized as a commensal that inhabits the intestinal tract of avian species as well as mammals. Since 2006, several outbreaks of spondylitis, vertebral abscesses, progressive lameness, and septicemia associated with E. cecorum in broiler breeders and straight-run broilers were reported from many countries including the United States.12,13,17 Increased mortality and high culling rates associated with the disease could be economically devastating to the industry. Because the route of entry, the mode of transmission, and the virulence mechanisms of E. cecorum infections are unknown, identification of the source of outbreaks is required for control and prevention. The objective of the current study was to assess the genetic polymorphism of clinical isolates of E. cecorum of diverse origins in order to understand their relatedness and to identify possible pathogenic clones using 3 molecular typing methods, namely PFGE, RAPD-PCR, and ERIC-PCR. Also, the typeability, discriminatory power, reproducibility of results, and feasibility of the technique of the each typing method were evaluated.
Both PFGE and ERIC-PCR methods demonstrated a high genetic variability in E. cecorum isolates. Interestingly, all 3 typing methods gave indistinguishable fingerprints for isolates 14 and 15 from company G with 100% similarity. These 2 isolates originated from infected birds on 2 different farms (farms G1 and G2) in outbreaks that occurred during the same month. This suggests that the source of E. cecorum outbreaks in farm G1 and farm G2 might be from a common source (company G). The same explanation would be applied to isolates 5 and 6 that originated from company E. Also, isolates 25 and 28 from farm J1, which were isolated from the same outbreak at different time points, produced identical fingerprints with all 3 typing methods. This implies that the outbreak on farm J1 was associated with the same clone of E. cecorum. Isolates 2 and 16 from farm B1 and isolates 12 and 13 from farm B2 were identified as different genotypes by all 3 typing methods, suggesting that more than one clonal type of E. cecorum was associated with these outbreaks.
It is evident that pulsotyping appears to be a promising method for genotyping E. cecorum because of its superior discriminatory power and reproducibility compared to RAPD-PCR and ERIC-PCR. However, it is a time-consuming, labor-intensive, cumbersome technique for routine clinical application. The requirement of expensive equipment and the lengthiness of the procedure are major disadvantages of PFGE over PCR-based methods. In the present study, a modification of a previously published protocol of PFGE 10 was followed, which entailed a relatively simple method requiring only 2 days to obtain fingerprint patterns. Although PCR-based methods are very easy to perform and relatively inexpensive compared to PFGE, the results are more difficult to analyze and interpret due to the appearance of multiple weak bands in the PCR profiles when repeated.
In conclusion, E. cecorum isolates used in the present study revealed genetic polymorphism as detected by all 3 molecular typing methods. Furthermore, genotypes indistinguishable by all 3 methods revealed genetic relatedness between certain isolates, which could be rationalized by the source of infection and nature of outbreaks. Nevertheless, PFGE, RAPD-PCR, and ERIC-PCR techniques used in the present study for typing of E. cecorum revealed differences in terms of discriminatory power, reproducibility, ease of performance, and interpretation of results. Pulsed-field gel electrophoresis provided the most reliable results with greater discriminatory power and higher reproducibility compared to the 2 PCR-based methods. Although the discriminatory power of ERIC-PCR was not statistically different from that of PFGE, reproducibility was relatively low. The RAPD-PCR gave the lowest discriminatory power among the 3 typing methods. Further studies have to be undertaken with a larger number of isolates comprising both clinical and commensal E. cecorum collected from diverse sources for further evaluation of PFGE as a useful molecular typing method for epidemiological surveillance of E. cecorum outbreaks.
Footnotes
a.
Sensititre Gram-positive identification system, TREK Diagnostic Systems Inc., Cleveland, OH.
b.
Remel Inc., Lenexa, KS.
c.
Key Scientific Products Inc., Stamford, TX.
d.
Fisher Scientific, Pittsburg, PA.
e.
G-Biosciences, Maryland Heights, MO.
f.
Sigma-Aldrich, St. Louis, MO.
g.
Promega Corp., Madison, WI.
h.
Bio-Rad Laboratories, Hercules, CA.
i.
Alpha Innotech Corp., Santa Clara, CA.
j.
TopTaq DNA Polymerase, Qiagen Inc., Valencia, CA.
k.
Seakem agarose, Cambrex Corp., East Rutherford, NJ.
l.
BioNumerics version 4.0, Applied Maths Inc., Austin, TX.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Pennsylvania Department of Agriculture and Broiler Check-off Program of the Pennsylvania University.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.