
Abstract
Calf diarrhea (scours) is a primary cause of illness and death in young calves. Significant economic losses associated with this disease include morbidity, mortality, and direct cost of treatment. Multiple pathogens are responsible for infectious diarrhea, including, but not limited to, Bovine coronavirus (BCV), bovine Rotavirus A (BRV), and Cryptosporidium spp. Identification and isolation of carrier calves are essential for disease management. Texas Veterinary Medical Diagnostic Laboratory current methods for calf diarrhea pathogen identification include electron microscopy (EM) for BCV and BRV and a direct fluorescent antibody test (DFAT) for organism detection of Cryptosporidium spp. A workflow was developed consisting of an optimized fecal nucleic acid purification and multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) for single tube concurrent detection of BCV, BRV, and Cryptosporidium spp., and an internal control to monitor nucleic acid purification efficacy and PCR reagent functionality. In “spike-in” experiments using serial dilutions of each pathogen, the analytical sensitivity was determined to be <10 TCID50/ml for BCV and BRV, and <20 oocysts for Cryptosporidium spp. Analytical specificity was confirmed using Canine and Feline coronavirus, Giardia spp., and noninfected bovine purified nucleic acid. Diagnostic sensitivity was ≥98% for all pathogens when compared with respective traditional methods. The results demonstrate that the newly developed assay can purify and subsequently detect BCV, BRV, and Cryptosporidium spp. concurrently in a single PCR, enabling simplified and streamlined calf diarrhea pathogen identification.
Introduction
Neonatal calf diarrhea (calf scours) is a common disease in young calves. The clinical signs include diarrhea (watery yellow, gray, or greenish containing blood or mucus), depression and weakness, dehydration, weight loss, convulsions, and sometimes death upon disease progression. 5 The disease is complex and associated with multifactorial risk factors, such as environmental and management conditions, nutritional and immune status, and pathogen load. 4 Significant economic losses associated with this disease include morbidity, mortality, direct cost of treatment, and long-term effects on health and productivity of surviving calves.
Multiple pathogens are associated with calf diarrhea, which include, but are not limited to, Bovine coronavirus (BCV), bovine Rotavirus A (BRV), Cryptosporidium ssp., enterotoxigenic Escherichia coli, Salmonella enterica, and Giardia intestinalis.3,5,25–27 The reported prevalence of infections varies due to geographic locations, production type (dairy or beef), calf age, study population selection criteria, and sensitivity and specificity of the diagnostic tests used for detection.
The most recognized pathogens causing calf diarrhea are BCV, BRV, and Cryptosporidium ssp.4,14 Bovine coronavirus infection occurs commonly in calves 0–30 days old, and infection rates of 7–16% in samples of diarrhea have been reported.4,5,7,14,19,23,25,28 Bovine Rotavirus A, initially known as Neonatal calf diarrhea virus, was one of the first identified viral causes of diarrhea. 14 There are at least 7 rotavirus antigenic groups (A–G), with groups A–C found in mammals. 13 However, rotavirus group A strains are among the enteropathogenic agents more commonly associated with neonatal diarrhea in calves up to 30 days old. 17 Infection appears and spreads rapidly causing extensive damage to the intestinal lining which results in rapid fluid loss and dehydration5,14; BRV infection rates of 20–60% in samples of diarrhea have been reported.1,6,15,25 Another noted intestinal pathogen is the protozoan Cryptosporidium ssp. (particularly Cryptosporidium parvum), which causes infection in calves 3–21 days of age but rarely occurs after 3 months of age. Infection rates of 10–55% in samples of diarrhea have been reported.6,11,25 Calves infected with 2 or more pathogens are 6 times more likely to develop clinical diarrhea than those infected with a single pathogen. 7 Thus, the testing of diarrhea samples provides a valuable tool for etiologic diagnosis of calf scours. To this end, a streamlined workflow consisting of a nucleic acid purification protocol and multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) for the detection of calf diarrhea pathogens (BCV, BRV, and Cryptosporidium spp.) in fecal samples was developed and its performance evaluated.
Traditional methods (at Texas Veterinary Medical Diagnostic Laboratory [TVMDL], College Station, TX, and West Amarillo, TX) for calf diarrhea pathogen (CDP) identification include electron microscopy (EM) for BCV and BRV and the direct fluorescent antibody test (DFAT) for Cryptosporidium. To simplify and streamline the detection of these pathogens, a robust single-tube multiplex RT-qPCR assay with equivalent traditional method sensitivity for the detection of BCV, BRV, and Cryptosporidium in fecal samples was developed and optimized. The nucleic acid purification method utilized a workflow that is routinely used in the TVMDL for the purification of pathogens from diverse biological samples, thus calf diarrhea samples could be easily incorporated with other submitted diagnostic samples, minimizing labor and equipment. In addition, an artificial exogenous internal nucleic acid purification and amplification control was included in the assay in order to minimize false-negative results.
Materials and methods
Clinical specimens
A total of 130 fecal samples, submitted to the TVMDL and the Oregon State University Veterinary Diagnostic Laboratory (OSUVDL; Corvallis, OR) for clinical diagnosis were used to evaluate the performance of the multiplex RT-qPCR. All samples were from clinically diseased animals and were tested by both traditional test methods (EM for BCV and BRV, and DFAT for Cryptosporidium). Of the 72 samples in which the age of the animal was provided with sample submission, the median age range was 2 weeks of age.
Oligonucleotides
The oligonucleotides sequence information and the final reaction (1×) concentrations in the 25-µl RT-qPCR are provided in Table 1. Primers and probes sequences for detection of Cryptosporidium ssp. were adopted from a previous publication. 24 Bovine coronavirus and BRV nucleotide sequences available in GenBank (query March 2010) were sorted by gene and independently aligned using the MUSCLE software. 12 MUSCLE analysis identified the most conserved sequences in the BCV spike structural protein gene and the BRV nonstructural protein 5 gene. Subsequently, alignment using BCV spike structural protein gene sequences (n = 53) and complete genome sequences (n = 19) was used for BCV assay selection; alignment using BRV nonstructural protein 5 sequences (n = 60) was used for BRV assay selection. Primers and probes were positioned in conserved nucleotide sequences using primer/probe design software e and BLASTed against the National Center for Biotechnology Information “nucleotide (nr/nt)” database and Bos taurus genome to confirm specificity. The synthetic construct clone NISTag38 external RNA control sequence (GenBank accession no. DQ883679) is a unique artificial antigenomic sequence with no significant homology to annotated species sequences. This sequence was used as the exogenous internal positive control (XIPC) target. The XIPC primers and probe were generated using primer/probe design software. e All primers and probes were proprietary. f
Primers and probes for the calf diarrhea pathogen multiplex reverse transcription quantitative polymerase chain reaction assays.*
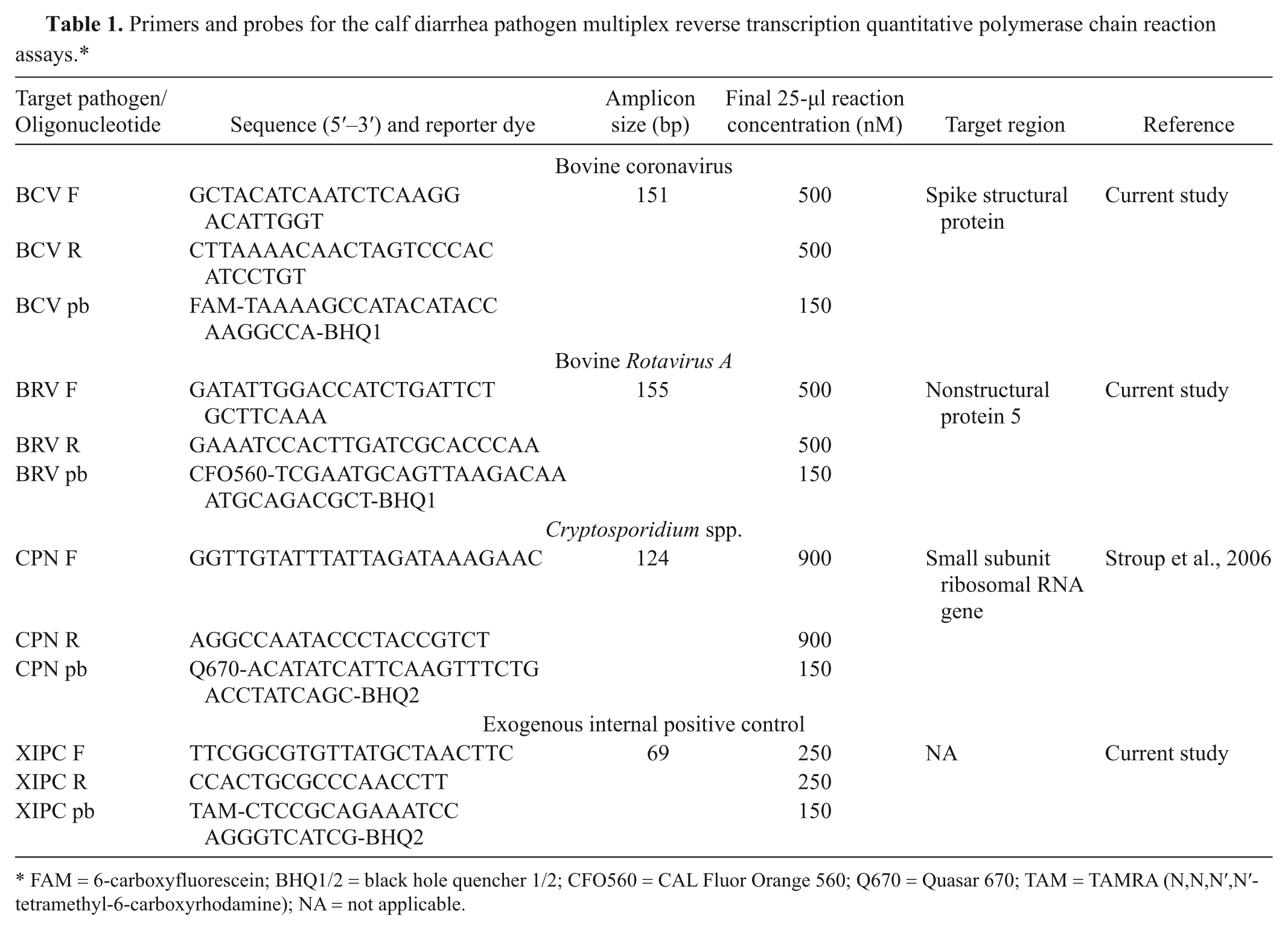
FAM = 6-carboxyfluorescein; BHQ1/2 = black hole quencher 1/2; CFO560 = CAL Fluor Orange 560; Q670 = Quasar 670; TAM = TAMRA (N,N,N′,N′-tetramethyl-6-carboxyrhodamine); NA = not applicable.
Plasmids
An in vitro transcribed CDP control RNA, containing a fusion of the target sequences of BCV, BRV, and Cryptosporidium spp., and an in vitro transcribed XIPC RNA was generated for multiplex RT-qPCR optimization. DNA sequences specific to CDP (i.e., BCV, BRV, and Cryptosporidium spp.) and XIPC were chemically synthesized and cloned into plasmid vectors. g Plasmids were linearized using HindIII restriction enzyme digestion and subsequently transcribed into RNA using a commercial transcription kit. e In vitro transcription was performed according to the manufacturer’s instructions. Amplicons were quantitated using a spectrophotometer h and sizes verified by gel electrophoresis. i
Nucleic acid purification
Nucleic acid was purified from all samples using a commercial RNA isolation kit and supplementary lysis/binding solution concentrate. e Briefly, fecal suspensions (approximately 0.1g of feces or 100 µl in 1-ml 1× phosphate buffered saline [PBS], pH 7.5–8.0) were mixed for 1 min at 15 Hz on a tissue lyser. i Fifty microliters of fecal suspension was mixed with 400 µl of lysis solution, e 1 µl of carrier RNA (1 µg/µl), and 1 µl of XIPC RNA (at 10,000 cp/µl) for 5 min at 15 Hz and centrifuged at 20,000 × g for 3 min to generate the clarified lysate. The clarified lysate (350 µl) was transferred to a 96-well, deep-well plate containing 20 µl of magnetic bead mix (10 µl of lysis/binding enhancer and 10 µl of RNA binding beads), and 200 µl of isopropanol was added; this plate was denoted as the sample plate. The following 96-well plates were loaded onto an automated magnetic particle processor h for nucleic acid purification: sample plate, wash solution 1 plate (300 µl/well), wash solution 2 plate (300 µl/well), and elution buffer plate (90 µl/well). The nucleic acid purification procedure consisted of the following steps: lysis/binding for 5 min, one 2-min wash 1, one 2-min wash 2, 1-min dry step, and a 3-min heated elution step at 70°C; detailed protocol program/script is provided in the online appendix. Purified nucleic acid was then denatured at 95°C for 5 min in a thermal cycler to denature the double-stranded BRV RNA for RT-qPCR.
Multiplex RT-PCR assay
The TaqMan multiplex RT-qPCR (25 µl total volume) utilized the components of a commercial RT-PCR kit e (according to manufacturer’s instructions) and the primers and probes for BCV, BRV, Cryptosporidium spp., and the XIPC positive control; the final concentration (per 25 µl) of each oligo is provided in Table 1. Each reaction contained 12.5 µl of 2× RT-PCR buffer, 2.5 µl of 10× enzyme mix, 1 µl of 25× primer probe mix (contained all oligos in Table 1), 1 µl of nuclease-free water, and 8 µl of nucleic acid template. The RT-qPCR was performed using a real-time PCR system. e The cycling conditions were as follows: reverse transcription at 48°C for 10 min (single cycle), activation/denaturation at 95°C for 10 min (single cycle), and 40 cycles of amplification at 95°C for 15 sec and 60°C for 45 sec. Samples with a quantification cycle (Cq) ≤37.0 cycles were considered positive.
Electron microscopy for the detection of Bovine coronavirus and bovine Rotavirus A
Two different procedures were used by the TVMDL and the OSUVDL, respectively, for EM. For the TVMDL EM, approximately 5 g of feces was resuspended in 10–15 ml of PBS and centrifuged at 2,500 × g for 15 min. The clarified supernatant (5 ml) was then centrifuged at 30,000 × g for 1 hr, and the resultant pellet was resuspended in 250 µl of water. Five drops of sample was mixed with 10 µl of water, 5 drops of phosphotungstic acid (pH 6.8), and 1 drop of 1% bovine serum albumin. Afterward, samples were put into a glass nebulizer and sprayed onto a 300-mesh grid. Bovine coronavirus and BRV were detected by examination under a microscope at 30,000× magnification.
For the OSUVDL EM, approximately 5 g of feces was resuspended in 25 ml of PBS and centrifuged at 5,000 × g for 20 min. The clarified supernatant (12 ml) was centrifuged at 100,000 × g for 1 hr, and the resultant pellet was resuspended in 1 ml of water. Five microliters of sample was mixed with 5 µl of 2% phosphotungstic acid (pH 6.9), and 1 µl of the mixture was then applied to the center of a 300-mesh grid. The sample was side blotted with a piece of torn filter paper to remove the majority of the sample, thus leaving a fine layer to air dry. Samples were examined under a microscope at 30,000× magnification.
Direct fluorescent antibody test for the detection of Cryptosporidium spp
All 130 samples (from the TVMDL and the OSUVDL) were examined for Cryptosporidium oocysts at the TVMDL using a commercial test kit j to ensure that a uniform protocol was used for detection of Cryptosporidium spp. Testing was done according to the manufacturer’s instructions. Briefly, fecal samples were smeared onto treated slides and allowed to dry. One drop of detection reagent and counter stain was added to each test well and control wells. Slides were incubated for 30 min in a humidified chamber and then washed with buffer. Mounting medium and coverslips were placed onto each slide. Slides were examined for characteristic oocyst morphology using a microscope with epifluorescence. A positive reaction consisted of brilliant fluorescence of Cryptosporidium oocysts against a dull reddish background; oocysts were visible as round to slightly oval with bright apple green–stained cell walls.
Statistical analysis
Repeatability of the multiplex RT-qPCR was determined by calculation of the intra- and interassay coefficient of variation (CV) for each pathogen assay. Intra-assay variability was analyzed in triplicate PCR at each dilution, and interassay variability was analyzed in 3 separate experiments (runs). Intra-assay CV = ([average of each run’s Cq standard deviation (SD)/average of each run’s Cq average] × 100); interassay CV= ([SD of the averages of each run/average of the averages of each run] × 100). Probit analysis was performed using statistical software k to assess the limit of detection of each assay.8,22 The PCR efficiency was calculated using the following formula: E = 10(–1/slope) − 1 × 100.
Performance of the multiplex RT-qPCR panel was evaluated by comparison against the traditional test methods (EM and DFAT). Cohen kappa and percentage agreement were calculated using a previously published spreadsheet. 18 It was expected that the newly developed CDP PCR assays had at least equal or higher diagnostic validity when compared to the traditional diagnostic methods, which precluded the use of DFAT and EM as the gold standard reference tests. Therefore, to obtain estimates of the diagnostic sensitivity and specificity of the CDP PCR assays, a latent-class statistical method was used. The “TAGS” algorithm (“Test in the Absence of a Gold Standard”) was implemented in the R software (http://www.r-project.org/). 20
Results
Calf diarrhea pathogen assays analytical specificity and sensitivity
Each CDP assay was evaluated by RT-qPCR to determine analytical specificity (Supplementary Fig. 1). The BCV assay detected available BCV strains, as well as the vaccine strain (Hansen isolate), d but did not detect Canine or Feline coronavirus. The BRV assay also detected available BRV strains, vaccine strains (Lincoln isolate, serotype G6; B223 isolate, serotype G10), d and porcine Rotavirus A. The Cryptosporidium assay detected C. parvum and C. muris, but not Giardia intestinalis, Giardia lamblia, or Giardia muris, a protozoan that has been reported in calf diarrhea fecal samples. All assays were specific for each CDP.
The analytical sensitivity of the multiplex RT-qPCR assay was determined using serial dilutions of an in vitro transcribed CDP control RNA, and XIPC RNA (100 copies/rxn; Fig. 1). The BCV and BRV assays detected less than 20 copies and the Cryptosporidium assay less than 50 copies of CDP control RNA; correlation coefficients for each assay ranged from 0.997 to 0.999. Analytical sensitivity of the multiplex RT-qPCR assay was compared to the respective single-plex RT-qPCR using pooled/mixed serial dilutions of each pathogen-purified nucleic acid. Each pool/mixture contained the respective complete pathogen genomic sequences, which mimicked the presence of multiple pathogens in diagnostic samples. Each PCR contained all 3 pathogen-purified nucleic acids at specified dilutions, which spanned the equivalent dynamic range; each PCR also contained 500 copies of XIPC RNA. The analytical sensitivity of the multiplex (mp) RT-qPCR was equivalent to that of the respective single-plex (sp) RT-qPCR (Fig. 2). The PCR efficiencies were determined to be as follows: sp BCV = 102.40% (R2 = 0.990), mp BCV = 108.12% (R2 = 0.993), sp BRV = 96.77% (R2 = 0.999), mp BRV = 97.26% (R2 = 0.999), sp Cryptosporidium = 99.31% (R2 = 0.996), mp Cryptosporidium = 97.94% (R2 = 0.999). Gel electrophoresis data of the single-plex and multiplex RT-qPCR is provided in Supplementary Figure 2. Analytical sensitivity was also assessed using serial dilutions of each respective pathogen-purified nucleic acid in the presence of the 2 other pathogen-purified nucleic acids at high quantity (i.e., Cq: 20–23); this reaction mimicked a diagnostic sample that contained 1 pathogen at varying titers/loads in the content of the other 2 pathogens at high titers/loads. The results showed that each assay of the multiplex RT-qPCR produced equivalent detection sensitivity in the presence of 2 high pathogen loads and equivalent pathogen loads (i.e., pooled mixture of the 3 pathogen-purified nucleic acid–specific serial dilutions, denoted as “3 CDP NA dilns”). Figure 3 displays the results for BCV. The Cq shift between “BCV NA dilns” and “3 CDP NA dilns” was a result of the pooling (1:1:1, 3-fold dilution) of each respective pathogen-purified nucleic acid serial dilution, which theoretically results in approximately 1.6 Cq difference; similar results were obtained for BRV and Cryptosporidium (data not shown).
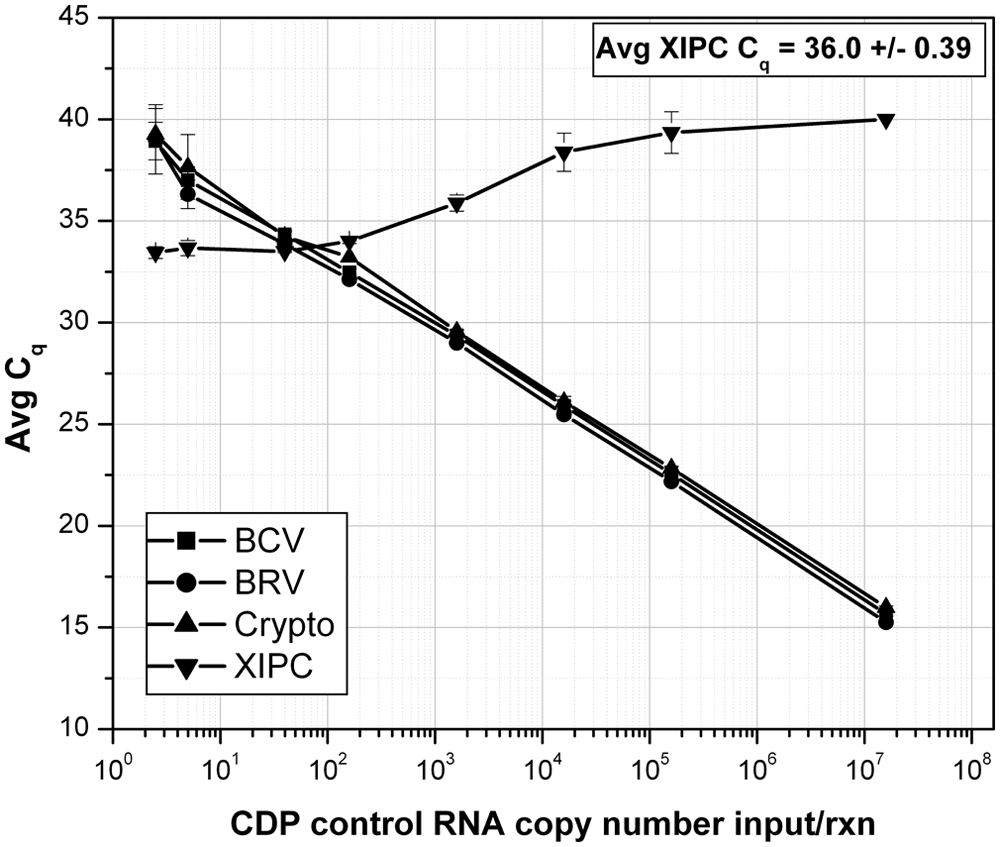
Analytical sensitivity of the calf diarrhea pathogen (CDP) multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) using in vitro transcribed fusion control RNA. Serial dilutions of in vitro transcribed CDP control RNA were prepared and amplified by RT-qPCR; n = 3 for each dilution. Each reaction contained 100 copies of exogenous internal positive control (XIPC) RNA. BCV = Bovine coronavirus; BRV = bovine Rotavirus A; Crypto = Cryptosporidium spp.; Cq = quantification cycle.
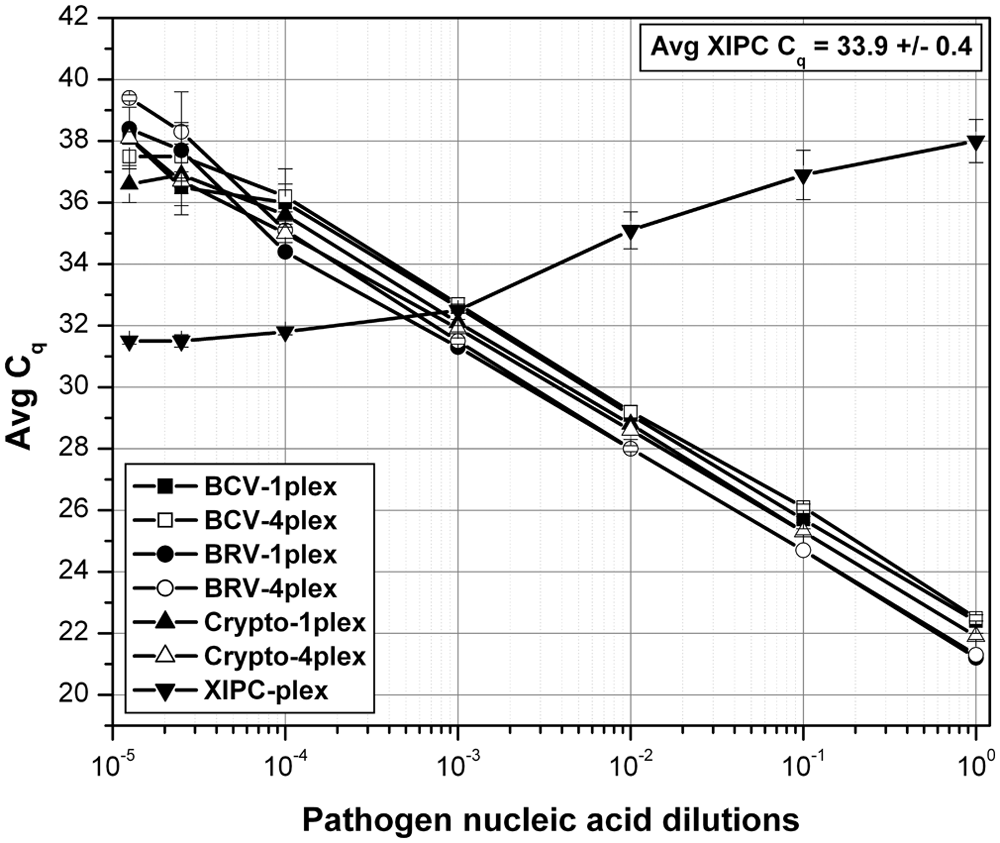
Analytical sensitivity of single-plex and multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) using serial dilutions of pooled purified pathogen nucleic acid. Serial dilutions of purified nucleic acid of each pathogen (Bovine coronavirus [BCV], bovine Rotavirus A [BRV], and Cryptosporidium spp.), spanning equivalent dynamic range (stock, -10×, -100×, -1,000×, -10,000×, -40,000×, and -80,000×) were prepared and pooled at 1:1:1 (3-fold) and analyzed by RT-qPCR (n = 3 for each dilution); each multiplex RT-qPCR contained 500 copies of exogenous internal positive control (XIPC) RNA. Estimated pathogen quantity in the first dilution PCR (25-µl reaction) was approximately 1,000 TCID50 for BCV, approximately 900 TCID50 for BRV, and approximately 450 cysts for Cryptosporidium. 1plex = single-plex RT-qPCR, 4plex = multiplex RT-qPCR; Cq = quantification cycle.
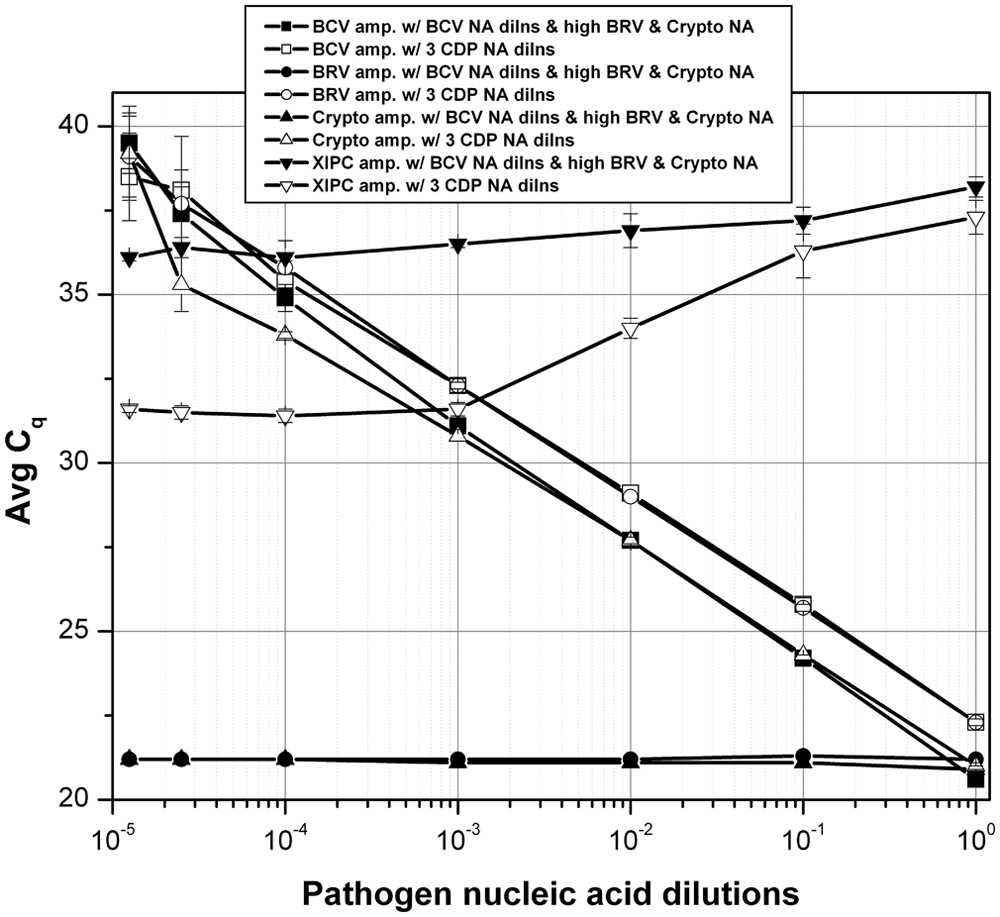
Analytical sensitivity of the calf diarrhea pathogen (CDP) multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) using serial dilutions of Bovine coronavirus (BCV)-purified nucleic acid (NA) and high quantity of bovine Rotavirus A (BRV)- and Cryptosporidium spp.–purified NA. Serial dilutions of BCV prepared as described in Figure 2 were prepared and analyzed by RT-qPCR. Each PCR contained a high quantity of BRV- and Cryptosporidium spp.–purified NA (i.e., quantification cycle [Cq] 20–23) and 500 copies of exogenous internal positive control (XIPC) RNA.
Each assay’s intra- and interassay repeatability was assessed at the specified pathogen-purified nucleic acid dilution in triplicate PCR in 3 separate experiments. The CV within runs (intra-assay variability) ranged from 0.3% to 3.7%, and between runs (interassay variability) ranged from 0.8% to 2.7% (Supplementary Fig. 3). Probit analysis, to determine the multiplex RT-qPCR detection rate of 95%, was also performed for each pathogen assay using the data set for the last 4 purified nucleic acid dilutions of the intra-and interassay variability experiment. For this analysis, a total of 9 reactions (responses) were used for each pathogen-purified nucleic acid dilution; Cq values below 40 were considered a positive response/amplification. A separate set of PCR targeting the CDP control RNA was performed to estimate copy number equivalents for the pathogen-purified nucleic acid dilutions. Probit analysis indicated that the multiplex RT-qPCR detection limit was less than 170 target copies per PCR for all 3 assays. Supplementary Figure 4 displays the probit plot for BCV (probability vs. log10 [dose] BCV-purified nucleic acid; 95% probit value of −0.56, estimated 50 BCV target copies per PCR). Similar plots were generated for BRV and Cryptosporidium spp. (data not shown).
Analytical sensitivity of the entire workflow (nucleic acid purification and detection methods) was determined using spiked fecal liquid samples with vendor-estimated concentrations of reference pathogen stocks (Fig. 4). Serial dilutions (n = 3 for each dilution) of a BCV, BRV, and Cryptosporidium spp. culture stock mix were prepared in CDP-negative fecal sample (10% w/v fecal suspension in 1× PBS). Nucleic acid was purified using the optimized nucleic acid purification method and analyzed by CDP multiplex RT-qPCR. The XIPC RNA (10,000 copies) was included in each purification to monitor purification efficiency and PCR inhibition. All 3 pathogens, and the XIPC RNA, were detected concurrently in all reactions. The Cq values of the XIPC RNA were consistent in all reactions, indicating effective nucleic acid purification and detection. The entire workflow estimated limit of detection (LOD) of each pathogen was determined as: BCV = 1.094 TCID50/ml, BRV = 0.875 TCID50/ml, and Cryptosporidium = approximately 10 oocysts. Correlation coefficients ranged from 0.944 to 0.999.
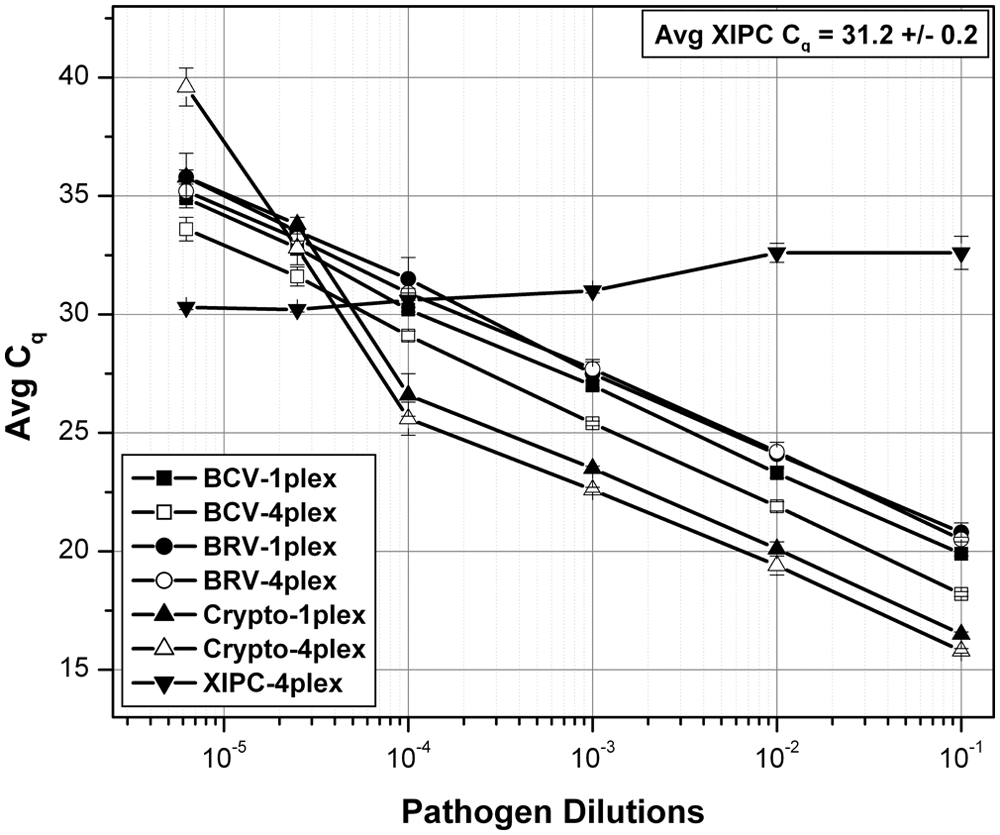
Analytical sensitivity of the calf diarrhea pathogen (CDP) nucleic acid purification and detection workflow using pathogen-spiked fecal samples. Serial dilutions of Bovine coronavirus (BCV), bovine Rotavirus A (BRV), and Cryptosporidium spp. culture stock mix were prepared in CDP-negative fecal liquid suspension (n = 3 for each dilution); dilution ranges were stock, -10×, -100×, -1,000×, -4,000×, and -1,600×. Nucleic acid was purified using the optimized nucleic acid purification method (10,000 copies of exogenous internal positive control [XIPC] RNA were spiked into each reaction) and analyzed by single-plex reverse transcription quantitative polymerase chain reaction (RT-qPCR) and CDP multiplex RT-qPCR. Cq = quantification cycle.
Determination of diagnostic sensitivity and specificity
A total of 130 calf diarrhea samples (with clinical signs), submitted to the TVMDL and OSUVDL for diagnostic testing, were used to evaluate the performance of the multiplex CDP RT-qPCR assay. Of those 130 samples, 10 (8%) were BCV positive, 44 (34%) were BRV positive, and 47 (36%) were Cryptosporidium positive by EM and DFAT, respectively. Fifty-two samples (40%) out of the 130 tested negative for all 3 pathogens via these traditional test methods.
All 130 fecal samples were evaluated by the multiplex CDP RT-qPCR assay to determine diagnostic sensitivity and specificity. Detection of the XIPC in all samples (average Cq: 31.4 ± 1.4) indicated effective nucleic acid purification and amplification. Results of the CDP assay were then evaluated against the traditional diagnostic testing methods (EM for BCV and BRV, and DFAT for Cryptosporidium; Table 2). The multiplex CDP RT-qPCR assay detected a total of 31 (24%) BCV-positive, 94 (72%) BRV-positive, and 58 (45%) Cryptosporidium-positive samples. Compared to EM, the CDP assay detected 21 more BCV positives (Cq range: 15.4–36.7) and 50 more BRV positives (Cq range: 11.0–36.7); the early Cq for these samples supports the presence of pathogen nucleic acid (Supplementary Fig. 5). Agreement between the traditional EM diagnostic test and the RT-qPCR assay was calculated to be 84% (κ = 0.42) and 62% (κ = 0.33) for the BCV and BRV assays, respectively. Compared to the DFAT, the CDP assay detected 12 more Cryptosporidium positives (Cq range: 20.0–35.9; Table 2); 1 DFAT-positive sample was negative by RT-qPCR (this result was likely due to a low number of oocysts in the swab liquid suspension that was used for nucleic acid purification). A substantial agreement (90%, κ = 0.80) was observed between the 2 test methods.
Comparison of calf diarrhea pathogen multiplex reverse transcription quantitative polymerase chain reaction (RT-qPCR) assay results with electron microscopy (EM) and a direct fluorescent antibody test (DFAT) for the detection of Bovine coronavirus (BCV), bovine Rotavirus A (BRV), and Cryptosporidium spp.
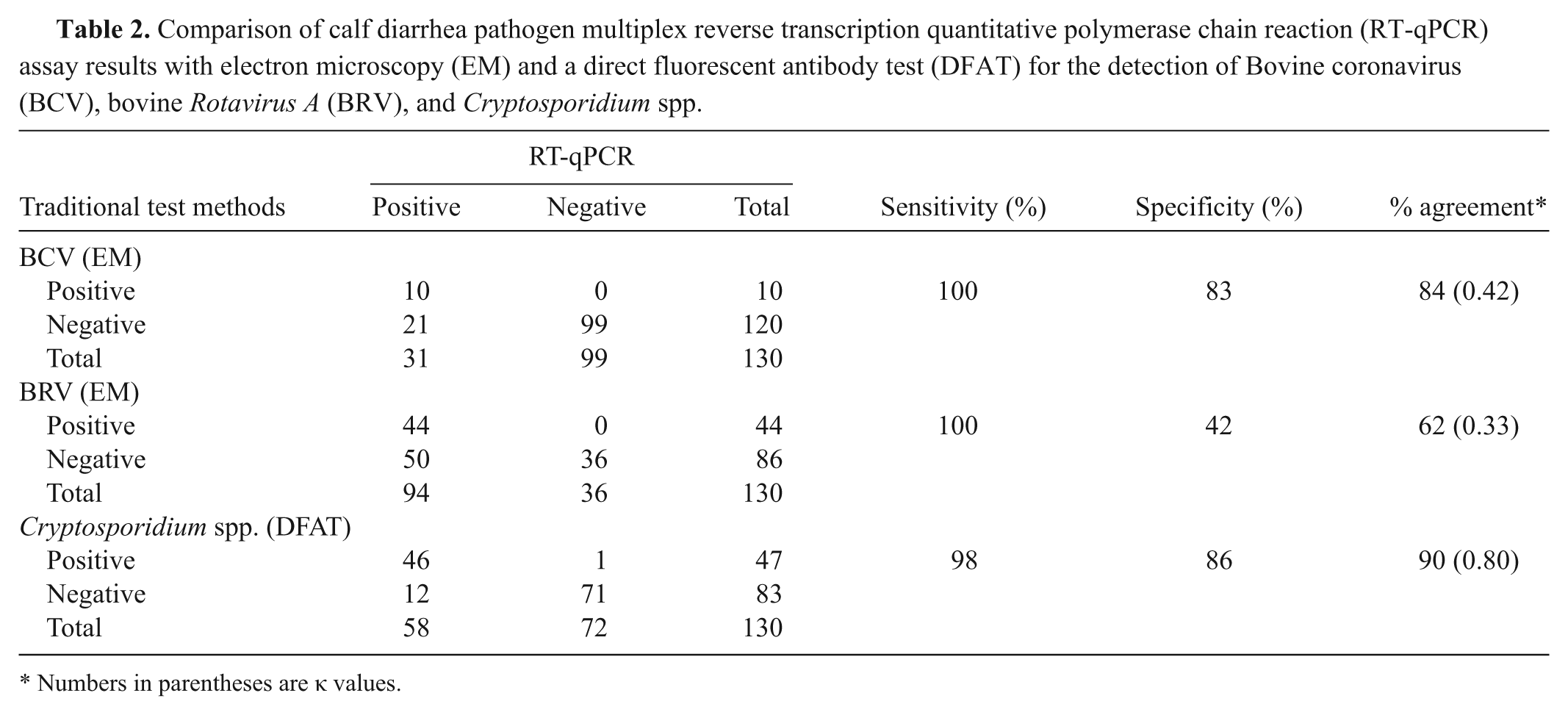
Numbers in parentheses are κ values.
Discussion
Calf scours causes a significant economic burden to producers due to morbidity, mortality, and treatment cost of diseased animals. The current TVMDL procedures for the diagnosis of the most recognized causative agents (BCV, BRV, and Cryptosporidium spp.) require multiple methods (EM and DFAT). Thus, the objective of the current study was to develop a simple and streamlined workflow for the detection of these pathogens. The workflow consisted of a nucleic acid purification method that is routinely used at TVMDL for the purification of various pathogens from diverse biological samples. This approach enabled diagnostic samples to be batched and provided cost and labor efficiencies. The purified nucleic acid was analyzed by a multiplex calf diarrhea pathogen RT-qPCR assay for the concurrent detection of BCV, BRV, and Cryptosporidium spp. in a single tube. Compared to the internal traditional diagnostic test methods currently used (EM and DFAT), the calf diarrhea pathogen assay was more simplified and streamlined; samples were processed using 1 protocol in less than 4 hr, enabling quick turnaround time with minimal reagents and equipment investment.
In comparison to the internal traditional EM and DFAT methods currently used, the diagnostic sensitivity of the calf diarrhea pathogen assay was 100% for BCV, 100% for BRV, and 98% for Cryptosporidium spp., respectively. Discrepancies between the traditional methods and the multiplex assay were noted in several samples. For those samples that were negative by the traditional methods, but positive by RT-qPCR (n = 83), the discrepancy is likely attributable to the increased sensitivity of the RT-qPCR assay.
Both the BCV and BRV RT-qPCR assays were highly sensitive (100%), as all EM-positive samples were detected by PCR. There were a number of samples, however, that were positive for BCV (n = 21) and BRV (n = 50) by RT-qPCR, but negative by EM. Analysis of the Cq values for these samples revealed that a majority of them (17/21 for BCV and 46/50 for BRV) had values below 35 (Supplementary Fig. 5). Thus, these samples were likely true BCV and BRV positives that were not identified by EM. This observation is in agreement with the fact that EM is less sensitive than PCR, with an estimated limit of detection of 105–106 particles/ml. 21 Furthermore, EM results are dependent on the skill and expertise of the examiner, particularly the ability to distinguish viruses among a mixture of fecal debris. It is possible that these samples were EM false negatives due to an inability to clearly identify virus particles within this sample matrix. The low sensitivity and user skill associated with EM resulted in a diagnostic specificity of 83% and 42% for the BCV and BRV RT-qPCR assays, respectively.
Similarly, the Cryptosporidium RT-qPCR assay was more sensitive than the traditional test method; 12 DFAT-negative samples were RT-qPCR positive. Examination of the Cq values for these samples revealed that 11 out of the 12 had a Cq below 35.0, and thus were likely true positives (Supplementary Fig. 5). It is worth noting that 5 of the 12 discrepant samples were submitted as swabs. The use of swabs for sample collection can limit the detection of pathogens because pathogens can stick to the cotton fibers of the swab, and thus produce false-negative results. One DFAT-positive sample was RT-qPCR negative; follow-up analysis revealed a Cq of 37.5, which was above the established Cq cut-off of 37.0. Thus, the sample was considered to be negative by RT-qPCR.
Independent infection with BCV, BRV, and Cryptosporidium spp. was detected in 5%, 21%, and 1.5%, respectively, of the fecal samples as determined by RT-qPCR. Coinfections with BRV and Cryptosporidium spp. were found in 28%, BCV and BRV in 7%, and BCV, BRV, and Cryptosporidium spp. in 12% of the samples; no BCV and Cryptosporidium spp. coinfections were present. The BRV and Cryptosporidium spp. coinfections correlated with several reports describing the prevalence of concurrent infections with rotavirus and Cryptosporidium in calves with diarrhea.10,16,25 However, the extent to which these 2 pathogens, as well as coronavirus, are involved in causing disease is unknown. All samples submitted for diagnostic testing were from clinically diseased calves, yet 24 samples (18.5%) tested negative for all 3 pathogens using RT-qPCR. It is important to remember that the presence of a particular pathogen does not necessarily mean it is the cause of the diarrhea, as numerous pathogens are associated with causing diarrhea in calves. Therefore, while the multiplex assay may identify specific pathogens in diseased animals, the results must be carefully interpreted in terms of clinical diagnosis and treatment.
The inclusion of an internal control in the calf diarrhea pathogen assay assisted in identifying possible false negatives for each sample. Because the XIPC was amplified with every sample, effective nucleic acid purification and PCR reagents functionality were confirmed, hence decreasing the potential for false-negative results.
Several articles detail the development of RT-PCR assays for the detection of pathogens responsible for calf diarrhea.2,9,28 The RT-qPCR assay described herein differs by enabling simultaneous detection of viral and protozoan nucleic acid in a single reaction tube, rather than as 2 separate reactions. In addition, an internal control, included during the nucleic acid extraction step, monitors the nucleic acid purification and amplification efficacy.
In conclusion, a multiplex RT-qPCR assay was developed for the simultaneous detection of 3 pathogens (BCV, BRV, and Cryptosporidium spp.) that contribute to calf diarrhea. The assay consisted of a single protocol for detection of 3 pathogens, included an internal positive control to monitor nucleic acid purification and amplification efficiency, provided high sensitivity (98–100% for all 3 pathogens), and enabled quick turnaround time and cost efficiencies.
Footnotes
Acknowledgements
The authors are very grateful to Dr. Thomas Hairgrove for valuable technical support and samples procurement.
a.
Viruses and protozoan reference strains, American Tissue Culture Collection, Manassas, VA.
b.
Bovine rotavirus strains, National Veterinary Services Laboratory, Ames, IA.
c.
Cryptosporidium and Giardia isolates, Waterborne Inc., New Orleans, LA.
d.
Calf-Guard and ScourGuard 3(K), Pfizer Animal Health, Exton, PA.
e.
Primer Express 3.0 software, MEGAscript T7 High Yield Transcription Kit, MagMAX-96 Viral RNA Isolation Kit, MagMAX Lysis/Binding Solution Concentrate, 7500 Fast Real-Time PCR System; Applied Biosystems, Carlsbad, CA.
f.
Primers and probes, Biosearch Technologies, Novato, CA.
g.
pBluescript II SK+ plasmid vectors containing DNA sequences specific to BCV, bovine BRV, Cryptosporidium, and XIPC, Blue Heron Biotechnology, Bothell, WA.
h.
Nanodrop spectrophotometer, KingFisher 96 Magnetic Particle Processor; Thermo Fisher Scientific, Wilmington, DE.
i.
QIAxcel, Qiagen TissueLyser; Qiagen Inc., Valencia, CA.
j.
MERIFLUOR Cryptosporidium/Giardia test kit, Meridian Bioscience Inc., Cincinnati, OH.
k.
NCSS 2007 Software, NCSS LLC, Kaysville, UT.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.