
Abstract
Calf diarrhea is a major economic burden to the bovine industry. Since multiple infectious agents can be involved in calf diarrhea, and the detection of each of the causative agents by traditional methods is laborious and expensive, a panel of 2 multiplex real-time polymerase chain reaction (PCR) assays was developed for rapid and simultaneous detection of the 5 major bovine enteric pathogens (i.e., Bovine coronavirus [BCoV; formally known as Betacoronavirus 1], group A Bovine rotavirus [BRV], Salmonella spp., Escherichia coli K99+, and Cryptosporidium parvum). The estimated detection limit (i.e., analytic sensitivity) of the panel was 0.1 TCID50 (50% tissue culture infective dose) for BCoV and group A BRV; 5 and 0.5 colony-forming units for E. coli K99+ and Salmonella, respectively; and 50 oocysts for Cryptosporidium per reaction. In testing 243 fecal samples obtained from submissions to the Iowa State University Veterinary Diagnostic Laboratory or from experimental animals with known infection status, the newly developed multiplex realtime PCR panel simultaneously detected all 5 pathogens directly from fecal samples and was more rapid and sensitive than the traditional diagnostic tests. The PCR panel showed 89%–97% agreement with those conventional diagnostic tests, demonstrating diagnostic sensitivity equal to or better than that of the conventional tests. In conclusion, the multiplex real-time PCR panel can be a tool for a timely and accurate diagnosis of calf diarrhea associated with BCoV, group A BRV, E. coli K99+, Salmonella, and/or Cryptosporidium.
Keywords
Introduction
Calf diarrhea causes major economic losses to the bovine industry because of high mortality and morbidity. According to the 2007 National Animal Health Monitoring System for U.S. dairy (http://nahms.aphis.usda.gov/dairy/index.htm), more than 50% of deaths in unweaned calves were due to diarrhea. Although noninfectious factors, such as insufficient uptake of colostrum, poor sanitation, stress, and cold weather, could cause neonatal calf diarrhea, various infectious agents such as viruses, bacteria, and protozoa are involved in calf diarrhea. 25 The major infectious agents that have been implicated in calf diarrhea are Bovine coronavirus (BCoV), group A Bovine rotavirus (BRV), and Bovine viral diarrhea virus 1 and 2 as viral agents; Salmonella spp., Escherichia coli K99+, and Clostridium spp. as bacterial agents; and Cryptosporidium as a protozoan agent. Among these, BCoV, group A BRV, Salmonella spp., E. coli K99+, and Cryptosporidium parvum are known as the 5 most common pathogens identified in scouring calves less than 2 months of age. 1,23,24,26
Bovine coronavirus is an enveloped virus with positive-sense, single-stranded RNA genome and is classified into genus Betacoronavirus as species Betacoronavirus 1 along with Mouse hepatitis virus, Human coronavirus OC43, Rat sialodacryoadenitis virus, Porcine hemagglutinating encephalomyelitis virus, Canine respiratory coronavirus, and Equine coronavirus. 9,14 Besides causing calf diarrhea, BCoV infection is also associated with winter dysentery in adult cattle as well as respiratory disease in all ages of cattle. 7,18 Group A BRV is a nonenveloped virus and possesses 11 double-stranded RNA segments. 24 Most BRVs belong to group A rotaviruses based on the antigenic similarity of the intermediate capsid protein (VP6), which is responsible for 95% BRV infection in the world; although groups B and C rotaviruses have also been identified in the field cases of BRV infection. 8,12,19,28 Both BCoV and group A BRV are often detected concurrently in scouring calves. 4,24 Infection with the viruses reduces the absorptive capacity of the intestines because of destruction of enterocytes and disrupts reabsorption of water, thereby leading to loss of fluid and electrolytes. 24
The diarrhea caused by Salmonella infection is characterized by watery and mucoid diarrhea with the presence of fibrin and blood. 11 Even though Salmonella can cause diarrhea in both adult cattle and calves, infection is more common and often causes severe symptoms in 10-day to 3-month-old calves. 11 On the other hand, E. coli K99+ causes a watery diarrhea, dehydration, and weakness in 1- to 4-day-old newborn calves. 3 The fimbrial adhesion F5 (K99) promotes the attachment of bacterial cells to glycoproteins on the surface of epithelial cells of the jejunum and/or ileum, and bacterial enterotoxin also causes damage to the epithelial cells, resulting in fluid secretion and diarrhea. 2
In addition to the viral and bacterial agents described above, the protozoan parasite, Cryptosporidium, also causes severe acute diarrhea in various animal species. 5 Cryptosporidium parvum is the most important protozoa related to calf diarrhea in 1- to 4-week-old calves. 26 The invasion of Cryptosporidium in enterocytes induces changes in the cell eytoskeleton, including the absence of microvilli and the shortening of columnar epithelial cells. These changes greatly contribute to the development of diarrhea due to malabsorption and fermentation of undigested milk in the intestinal lumen of young calves. 10
Since these various pathogens are individually or concurrently involved in calf diarrhea, differential diagnosis of these pathogens with rapid turnaround time is essential to implement appropriate treatment and preventive practice in a timely manner. Laboratory techniques that have been commonly used in veterinary diagnostic laboratories to identify these pathogens are virus isolation, electron microscopy, bacterial culture, fecal flotation method, antigen-capture enzyme-linked immunosorbent assay (Ag-ELISA), latex agglutination test (LAT), and/or polymerase chain reaction (PCR). These conventional methods are laborious, expensive, and/or slow in turnaround. Furthermore, they can be relatively insensitive depending on the quality and timing of sampling. As rapid detection of these pathogens at the early stage of outbreak substantially contributes to minimizing the spread of infection and increasing treatment efficiency, a panel of 2 multiplex real-time PCR assays (hereafter, multiplex PCR panel), which can simultaneously detect the major causative pathogens (i.e., BCoV, group A BRV, Salmonella spp., E. coli K99+, and Cryptosporidium), were developed; and their performance and utility in diagnosis of calf diarrhea were evaluated in the current study.
Material and methods
Reference strains of virus, bacteria, and parasites
The current study used the Nebraska strain of Bovine coronavirus a ; 3 different strains (NCDV, WC3, and B223) of group A BRV and 2 bacterial strains, E. coli K99+ (ATCC 31616) and Salmonella typhimurium (ATCC 14028) b ; and the Iowa strain of C. parvum. c These agents were used in assessing the analytic sensitivity of tests, in addition to serving as positive controls.
Specimens
Experimental specimens. Five calves were inoculated with 3 ml of C. parvum prepared at the rate of 500 oocysts/ ml. A total of 30 fecal samples collected from the 5 calves at 0, 4, 8, 12, 16, and 20 days post-challenge were kindly supplied by Dr. Jeffrey Knittel at Boehringer-Ingelheim Vetmedica Inc. (St. Joseph, Missouri).
Clinical specimens. A total of 243 feces or intestinal contents, which were collected from diarrheic calves and submitted to the Iowa State University Veterinary Diagnostic Laboratory (ISU VDL) in 2007, were used to evaluate the performance of the multiplex real-time PCR panel in comparison with other laboratory procedures routinely used at ISU VDL for the same target agents (i.e., real-time reverse transcription [RT]-PCR for BCoV, Ag-ELISA for rotavirus group A, bacterial culture and LAT for E. coli K99+, bacterial culture and serotyping for Salmonella, and microscopic observation with acid-fast staining for Cryptosporidia). In addition, 72 fecal samples collected from clinically healthy cattle at 3 dairy farms in Iowa were also used to validate the specificity of the multiplex real-time PCR panel.
Nucleic acid extraction
Nucleic acids of all target agents were simultaneously extracted from specimens by use of a commercial nucleic acid isolation kit d as described in the manufacturer's manual. In brief, 0.01 M phosphate buffered saline (pH 7.4) was added to each sample to make 30% fecal homogenates. After centrifugation for 1 min at 100 × g to pellet larger-size particles, 175 μl of the supernatant of each sample was added to a bead tube containing zirconia beads and 235 μl of lysis/binding solution. The bead tube was beaten at 20 Hz for 5 min with a commercial high-throughput sample disruptor. e After the beating process, the bead tubes were centrifuged at 16,000 × g for 3 min, and the supernatant was carefully transferred into clean microcentrifuge tubes. After another centrifugation at 16,000 × g for 6 min, 115 μl of the supernatant was transferred to a 96-well, deep-well microplate, f which contained 20 μl of paramagnetic beads, and 65 μl of 100% isopropanol. The deep-well microplate and 5 additional 96-well plates (VWR; 2 plates with 150-μl washing solution 1 per well, 2 plates with 150-μl washing solution 2 per well, and 1 plate with 50-μl elution buffer per well) were placed in a magnetic particle processor g for automated extraction process. The automated process consisted of lysis/binding step for 5 min; 2-time first washing step each for 90 sec; 2-time second washing step each for 2 min and 30 sec, respectively; dry step for 1 min; and, finally, the elution step for 3 min. The extracted total nucleic acids in the elution plate were stored in −80°C until used for PCR reaction.
The nucleotide information of primers and probes used for multiplex polymerase chain reaction.*

BCoV = Bovine coronavirus; fwd = forward; rev = reverse; BRV = Bovine rotavirus.
Oligonucleotides
The sequence information of primers and probes used in the multiplex real-time PCR panel are summarized in Table 1. Primers and probes for E. coli K99+, Salmonella, and Cryptosporidium were adopted from published information. The primers and probes for BRV were designed in the current study. Three each of forward and reverse primers and 2 probes were designed based on the VP6 gene (which encodes intermediate capsid protein), using Primer Express software h to cover all group A BRV strains, whose VP6 sequences are available in GenBank (Fig. 1). All of the primers and probes were synthesized and purchased from Integrated DNA Technologies (IDT), i except Minor Groove Binder probes for BCoV and BRV. d
Multiplex real-time PCR panel
The multiplex PCR panel was optimized with multiplex RT-PCR kit d following manufacturer's recommended protocol in a 25-μl reaction volume using 8 μl of extracted template. All primers and probes were prepared at 25-μM working concentration, and equal volumes of primers and probes were mixed together for each target agent. Two primers and 1 probe were mixed in a single tube for BCoV, E. coli K99+, Salmonella, or Cryptosporidium (i.e., BCoV mix, K99 mix, Salmonella mix, and Cryptosporidium mix, respectively), while 3 each of forward and reverse primers and 2 probes were mixed together for group A BRV (i.e., BRV mix). Two PCR reactions were prepared: one for viral agents (BCoV and BRV) and the other for bacterial/protozoan agents (E. coli K99+, Salmonella, and Cryptosporidium). The final concentration of each primer or probe was 0.2 μM. The PCR amplification was performed on an automated realtime PCR system. d Cycling conditions were as follows: (a) reverse transcription for 10 min at 45°C (omitted for bacterial/protozoan PCR); (b) a 10-min activation step at 95°C; and (c) 35 cycles of 15 sec at 95°C and 60 sec at 60°C. Samples with a threshold cycle (Ct) of 35 cycles or less were considered positive.
Internal control plasmid
Full-length genomic DNA (1768 base pair [bp]) of Porcine circovirus-2 (PCV-2), which has been detected only in swine, was amplified with PCV-2–specific primers, which contain XhoI and BamHI enzyme sites (Xho-PCVF0 5′-CTCGAGCTCGAGACCAGCGCACTTCGGCAGC-3′ and BamH-PCVR1768 5′-GGATCCGGATCCAATACT-TACAGCGCACTTCTTTCG-3′). The amplified PCR product was cloned into a pSK (+) vector j using the incorporated restriction enzyme sites. Based on the information of PCV-2 real-time PCR described previously, 22 5 nucleotides in the probe recognition site of the cloned PCV-2 genome were substituted with the sequences that have not been identified in any of PCV-2 isolates using a site-directed mutagenesis kit. j The sequence information of primers and probe for the internal control is described in Table 1. The internal control was added to either samples (before extraction) or extracts (after extraction) at a predetermined concentration, so that it could be detected between 32 and 34 cycles of amplification.
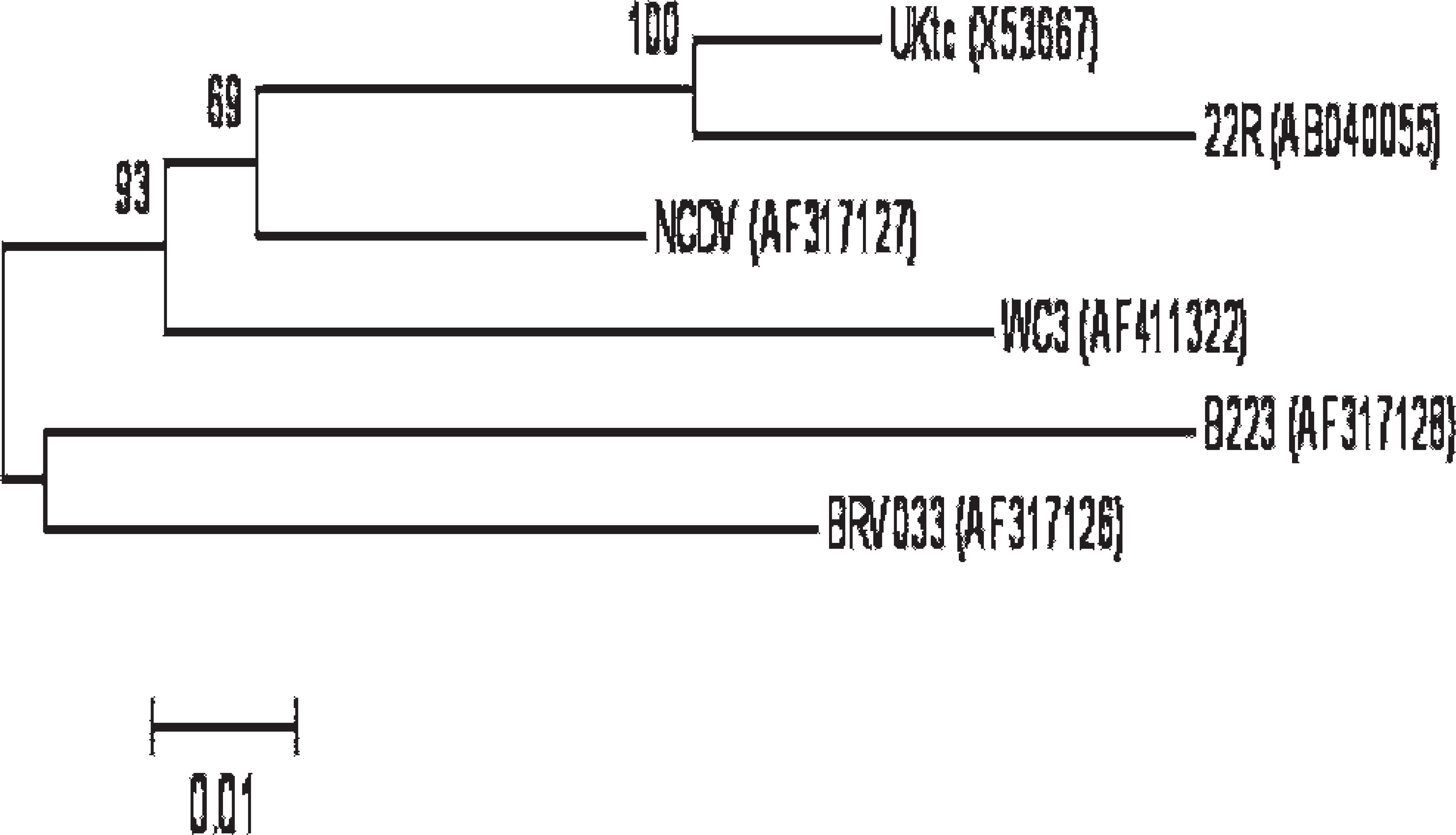
Genetic variation of VP6 genes among group A Bovine rotavirus. Each virus is indicated with strain name and GenBank accession number. The phylogenetic tree was constructed by neighbor-joining method. The reliability of analysis was determined by 1,000 times repeated bootstraps.
Bovine coronavirus real-time RT-PCR
A real-time PCR, which is routinely used to diagnose calf diarrheic cases at the ISU VDL, was employed. The BCoV real-time PCR was originally designed based on a previously reported BCoV gel-based PCR. 7 The diagnostic performance of the real-time PCR had been validated and optimized by testing reference viruses and clinical samples in comparisons with the gel-based PCR 7 and BCoV AgELISA at the ISU VDL (Harmon, unpublished data, 2003). The PCR was performed according to the protocol that has been routinely used at the ISU VDL. RNA was extracted from specimens with a commercial viral RNA isolation kit d as described in the manufacturer's manual. The PCR was performed with a commercial probe RT-PCR Kit e in a 25-μl reaction volume using 5-μl extracted template. Primers and probe were the same as employed in the multiplex PCR (Table 1), and the final concentration of primers or probe was 0.4 or 0.2μM, respectively. The PCR amplification was performed on an automated real-time PCR system k as follows: 50°C for 30 min, 95°C for 15 min, and 35 cycles of 94°C for 15 sec and 60°C for 60 sec. Samples with a Ct of 35 cycles or less were considered positive.
Rotavirus group A Ag-ELISA
Samples were assayed by a microplate enzyme immunoassay 1 following the procedures recommended by the manufacturer. 20 Samples with optical density ≤0.3 at 450 mm were considered positive for rotavirus.
Isolation and identification of Salmonella species
Samples were inoculated in tetrathionate broth m and incubated for 24 hr at 42°C for enrichment. The enriched samples were plated on brilliant green agar with novobiocin, m Hektoen enteric agar, n and/or XLT4 agar, m and then incubated at 35–37°C for 24 hr. Suspect Salmonella were subcultured from brilliant green (pink colonies), Hektoen enteric (green or black colonies), or XLT4 (black colonies) and confirmed as Salmonella in Kligler, urea, sulfur-indole-motility (SIM), and lysine agars m and in a commercial Gram-negative identification panel. o Colonies were serogrouped with commercial antisera p at ISU VDL or serotyped at the NVSL (Ames, Iowa) for final identification.
Isolation and identification of Escherichia coli K99+
Samples were plated directly onto Tergitol 7 agar with triphenyltetrazolium chloride n and incubated aerobically at 35–37°C for 24 hr. Escherichia coli colonies with rough, intermediate, smooth, or mucoid morphology were subcultured to conventional biochemical tube media for identification. Kligler iron agar, SIM agar, and urea agar slants m were inoculated and incubated aerobically at 35–37°C for 24 hr. The reactions were read and then compared with charts to confirm the identification. Escherichia coli growth from direct plating or from pure culture plating to E-agar m was tested for the presence of K99 pilus antigen using an E. coli antigen test kit. q
Identification of Cryptosporidium by acid-fast staining
Each sample was smeared on a glass slide, and the smear was dried briefly in ambient temperature. The air-dried smear was fixed in methanol for 10 min and then placed in carbolfuchsin r for 1–2 hr. The smear was washed with tap water for 1 min and placed in 1% acid alcohol until no more red color ran off. After the smear was washed briefly with tap water, the smear was counterstained in 0.5% Fast Green for 1 min. The slide was read under a light microscope after brief steps of washing and drying. Red and halo-shaped oocysts were identified as cryptosporidia. 15
Analysis of discrepant test results
Samples with discrepant results between the multiplex real-time PCR and other traditional tests were retested by the multiplex PCR after re-extraction and further analyzed using sequencing and gel-based PCR for each target agent. In addition, reference BCoV, E. coli K99+ strains, or PCV-2 internal control plasmid were spiked in discrepant samples and tested again by the multiplex PCR to determine that negative PCR results were not due to PCR inhibition during the extraction or amplification procedures.
Sequencing of real-time PCR products. Real-time PCR products were electrophoresed and visualized on 2% gels to confirm the presence of specific amplified target gene with predicted molecular size. The amplicons were then purified using a PCR purification kit e and submitted to the ISU Nucleic Acid Facility for sequencing. The same amplification primers described in Table 1 were used for sequencing of each target agent. Nucleic acid sequences of the PCR products were aligned with known sequences of the corresponding agents and analyzed using Lasergene® MegAlign software. s
Gel-based PCR tests. Gel-based PCR for the 5 pathogens were optimized using a one-step RT-PCR kit. e The primers used to detect each agent are listed in Table 2. Cycling conditions of PCR were as follows: (a) reverse transcription for 30 min at 50°C; (b) a 15-min activation step at 95°C; (c) 40 cycles of 30 sec at 94°C, 60 sec at 55°C, and 60 sec at 72°C; and (d) final extension for 7 min at 72°C. Reverse transcription enzyme mix and reverse transcription step were omitted for the PCR reactions of E. coli K99+, Salmonella, and Cryptosporidium.
The nucleotide information of primer used for alternative gel-based polymerase chain reaction

Statistical analysis
The performance of the multiplex PCR was compared by calculating the percentage agreement with other test results and the K value as follows:
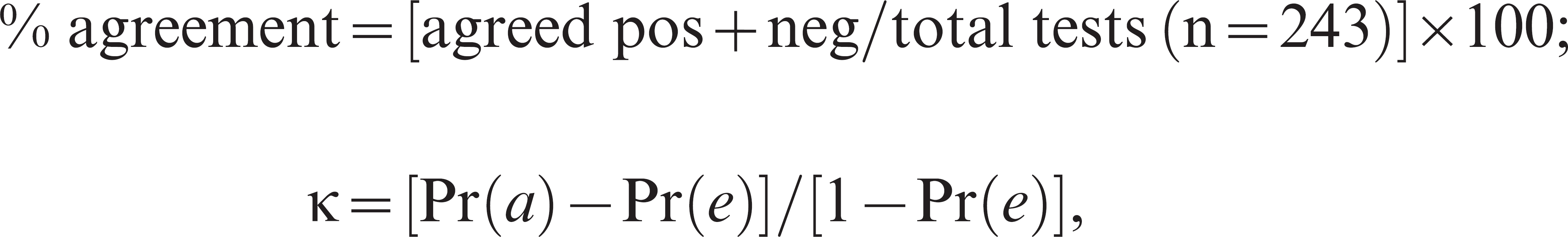
where Pr(a) is the relative observed agreement between tests, and Pr(e) is the probability that agreement is due to chance. If the tests are in complete agreement, then K = 1. If there is no agreement between the tests, then κ ≤ 0. The interpretation of K value was based on the guide provided previously 16 : poor agreement (κ = 0.00); slight (0.01 < κ < 0.20); fair (0.21 < κ < 0.40); moderate (0.41 < κ < 0.60); substantial (0.61 < κ < 0.80); almost perfect (0.81 < κ < 1.00).
Results
Optimization of multiplex real-time PCR panel
All reference strains of the 5 agents with known virus titer (50% tissue culture infective dose [TCID50]/ml), number of bacterial colony (colony-forming units [CFU]/ml), or number of oocysts (per ml) were serially diluted 10-fold and used to optimize the multiplex PCR. The multiplex PCR panel simultaneously detected all of the reference strains, yet only specific target agents without any false-positive result. Standard curves were generated using the 10-fold serial dilutions with correlation coefficients ranging from 0.987 to 0.996 and slopes of 3.06 to 3.85 (Fig. 2). The limits of detection (i.e., analytic sensitivity) for each agent are 0.1 TCID50 for BCoV and BRV; 5 and 0.5 CFU for E. coli K99+ and Salmonella, respectively; and 50 oocysts for Cryptosporidium per reaction.
As a next step, the performance of the multiplex PCR and simplex PCR for each of 5 agents was directly compared on the same 96 extracts to determine any negative effect of multiplexing on the PCR detection. The test results by both simplex and multiplex PCR were completely matched, and Ct differences between both PCR reactions were not statistically significant (P > 0.1), demonstrating that multiplexing did not cause significant negative effect on the sensitivity or specificity of the PCR reactions. In addition, 72 fecal samples collected from clinically normal cattle were tested by routine bacterial culture and the multiplex PCR panel. Only coliform bacteria were isolated from the culture, and all the samples were negative by the PCR panel for all 5 agents, suggesting that the PCR does not detect normal flora as any of the target agents.
When the multiplex PCR panel was run on 30 fecal samples collected from calves experimentally challenged with C. parvum, no amplification signal for Cryptosporidium was detected at 0 day post-challenge (dpc), and yet the highest number of oocyst (105.3–107.2 oocysts/ml) was detected in the fecal samples collected from the 5 calves at 4 or 8 dpc. At 20 dpc, 103.7 oocysts were still detected from 1 calf (Table. 3). No other agent (i.e., BCoV, group A BRV, E. coli K99+, Salmonella spp.) was detected from these fecal samples by the PCR panel.
Performance of multiplex PCR panel in comparison with other tests
Comparisons of test results on 243 scour samples between the multiplex PCR panel and other laboratory tests routinely used at ISU VDL for the 5 target agents are summarized in Table 4. Among all of the examined samples, the multiplex PCR panel detected the BCoV genome in 54 samples, which was 12 more than the number of positive samples identified by BCoV real-time RT-PCR (n 42), whereas all the other samples (n = 189) were negative for BCoV by both the PCR tests. Accordingly, the test agreement between BCoV RT-PCR and multiplex PCR was 95% (231/243), and K value was 0.844. In the case of BRV detection, the multiplex PCR identified 23 more samples as positive for BRV compared with the rotavirus Ag-ELISA, whereas 2 positive samples by the ELISA were negative for BRV by the multiplex PCR. Test agreement between the 2 tests was 89% (218/243), and K value was 0.733.
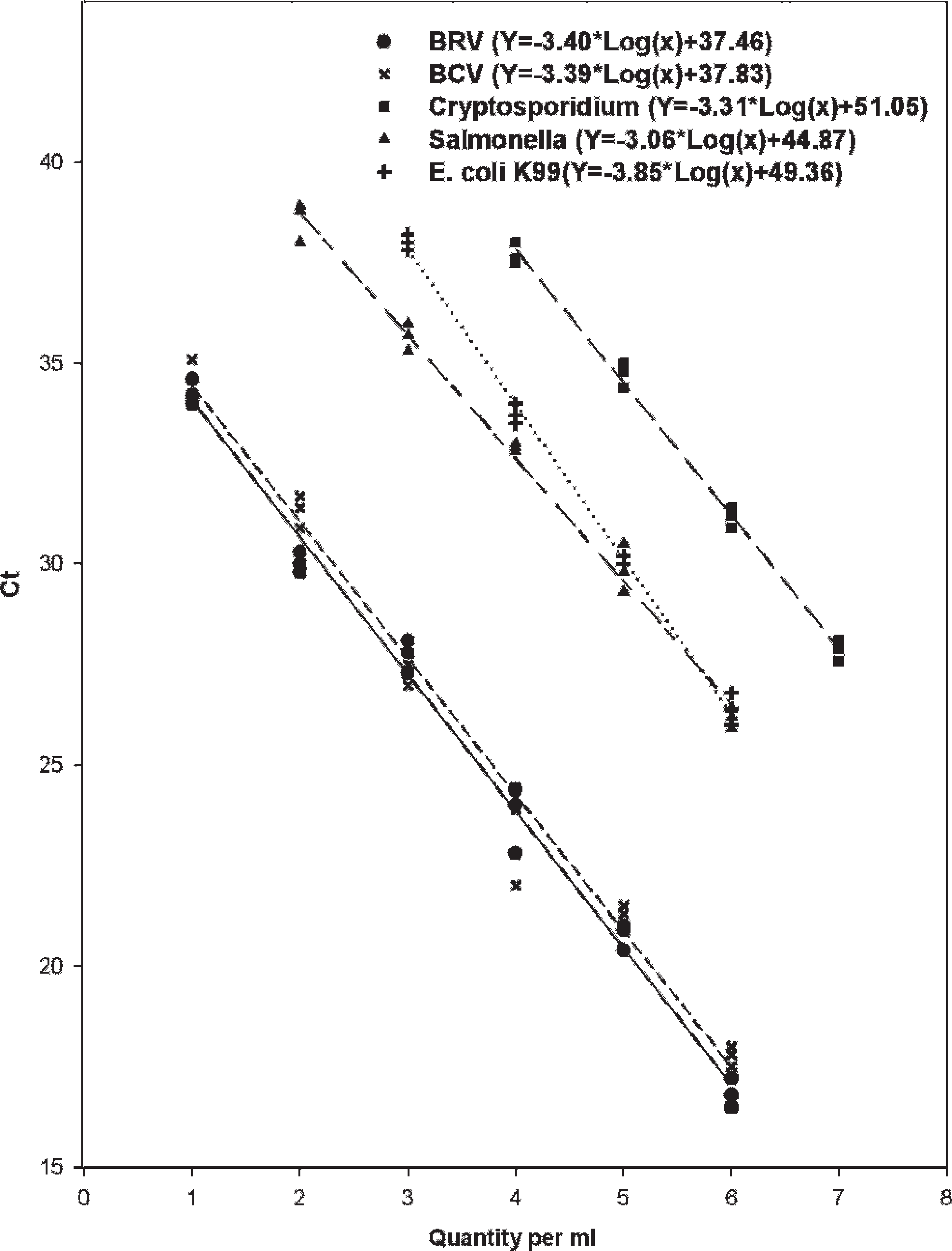
Multiplex detection of group A Bovine rotavirus (BRV), Bovine coronavirus (BCV), Salmonella sp., Escherichia coli K99+, and Cryptosporidium parvum. All of the 5 agents with known virus titer (50% tissue culture infective dose/ml), number of bacterial colony (colony-forming units/ml), or number of oocysts/ml were mixed and serially diluted 10-fold for simultaneous detection by multiplex polymerase chain reaction. Y-axis indicates cycle threshold (Ct) values. Each regression line was constructed based on 3 repeated measurements.
In comparison with bacterial culture methods for 2 bacterial pathogens (i.e., Salmonella and E. coli K99+), the multiplex PCR identified 4 more positive samples for Salmonella sp. compared with the culture results, whereas 3 positive samples by culture were negative by the multiplex PCR. The agreement between the multiplex PCR and Salmonella culture was 97% (236/243), and K value was 0.887. In the case of E. coli K99+, the multiplex PCR detected 39 positive samples, which were 9 more than the number of positive samples identified by culture and LAT. However, 5 positive samples by the culture method followed by LAT were negative by the multiplex PCR. Agreement of the 2 tests was 94% (229/243), and K value was 0.776.
The multiplex PCR was also compared with microscopic observation with acid-fast staining for the detection of Cryptosporidium spp. in the samples. The PCR detected 14 more positive samples than those by the microscopic observation, while 2 samples identified as positive for Cryptosporidium spp. by the microscopic observation were negative by PCR for C. parvum. Test agreement between the multiplex PCR and microscopic observation was 93% (227/243), and K value was 0.756.
Analyses of discrepant samples
All of the samples (n 12) that were negative by the multiplex PCR panel, but positive for any of the 5 target agents by other conventional tests (Table. 3), were retested by the multiplex PCR and gel-based PCR after re-extraction. The same results as initially observed were reproduced on both PCR assays. When reference BCoV, E. coli K99+ strains, or internal control plasmid at a known amount were spiked in those samples and tested, the spiked virus, bacteria, or internal control were detected as expected, confirming that the negative PCR results were not attributed to the presence of inhibitory substances in the samples. Nonetheless, the E. coli K99+ (n = 5) and Salmonella isolates (n = 3) were cultured from the samples, and all isolates were confirmed to be E. coli K99+ or Salmonella by the multiplex PCR.
For the samples (n = 62) that were positive for any of the target agents by the multiplex PCR, but negative by other conventional tests (BCoV [n = 12], BRV [n = 23], Salmonella [n = 4], E. coli K99+ [n = 9], or Cryptosporidium [n = 14]), the simplex PCR and gel-based PCR for each agent were repeated, revealing the same results as those by the multiplex PCR. Each of the PCR products had the expected molecular size for each agent (Fig. 3). The sequences from the PCR products shared 98–100% homologies with the target amplification regions for each agent.
Discussion
A multiplex real-time PCR panel that can simultaneously detect 5 major causative agents of calf diarrhea (i.e., BCoV, BRV, Salmonella, E. coli K99+, and C. parvum) was developed in the current study. As the new multiplex PCR can test 96 samples to determine the presence and absence of 5 different viral, bacterial, and protozoan agents within 4 hr, it can greatly reduce the cost, labor, and turnaround time compared with various routine diagnostic techniques, such as individual PCR, bacterial culture, serotyping, LAT, microscopic examination, and Ag-ELISA. Since fecal material is one of most difficult matrices from which to conduct PCR, and extraction is the most labor-intensive and expensive procedure in PCR performance, several innovative approaches were made in the multiplex PCR to decrease time, labor, and cost. First, the PCR employed a unique extraction method (i.e., total nucleic acid isolation kit), which can simultaneously purify either RNA or DNA of viral, bacterial, and protozoan agents directly from fecal materials in a single tube. Second, a magnetic particle processor g in a 96-well format was used in the extraction process to minimize inhibition problem from feces and make the test fit for high throughput, thus substantially reducing the extraction time (<1.5 hr for 96 samples). Third, the PCR reaction was done using one-step PCR procedure and in a fast gene amplification system, d which permits PCR to be completed within 2.5 hr. Any of these methods is applicable individually or in combination to development of other real-time PCR-based multiplex or panel testing, which becomes a common practice since disease problems in modern animal agriculture tend to be multifactorial.
Detection of Cryptosporidium parvum by multiplex polymerase chain reaction performed on fecal samples collected from calves experimentally challenged with C. parvum at 500 oocysts/ml.
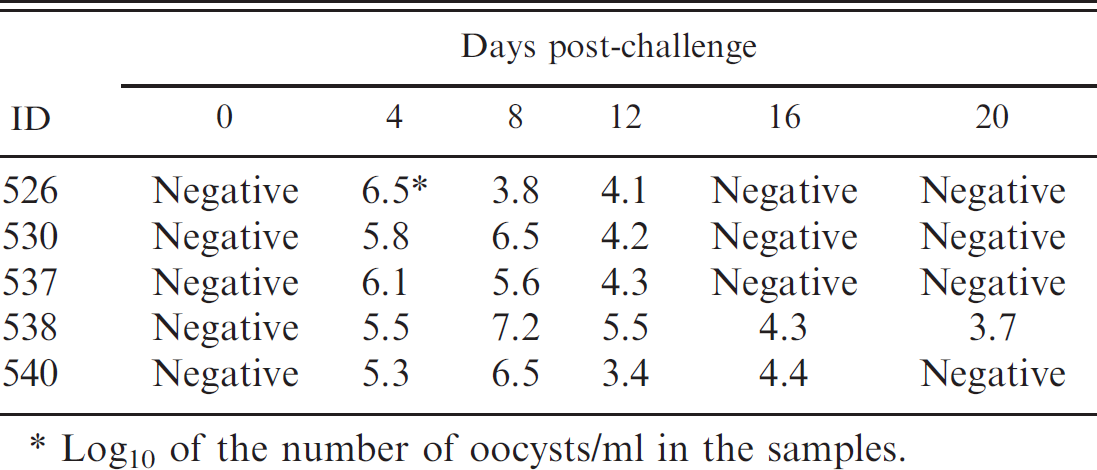
Log10 of the number of oocysts/ml in the samples.
Overall, a good agreement in the test results was observed between the multiplex PCR panel and the traditional diagnostic methods, ranging between 89% and 97% based on the test results of 243 clinical samples. Most of the discrepant results between the multiplex PCR and the traditional tests (62/74 total discrepant samples) were due to the higher sensitivity of the multiplex PCR panel, since the positive results of 62 samples for either of the 5 agents by the PCR panel could be confirmed with respective gel-based PCR tests or sequencing of the PCR products. The other 12 discordant samples, which were positive by the traditional tests, but negative by the multiplex PCR panel, were still negative by the respective gel-based PCR tests, suggesting that there was no detectable target gene in the samples. Reference BCoV, E. coli K99+ strain, or internal control plasmid spiked in those samples were successfully detected by the PCR panel, ensuring that the negative PCR results were not due to gene degradation or PCR inhibition during the extraction or amplification procedures. The possible explanations for the negative result of the multiplex PCR on these samples include the following: (a) false-positive results of the traditional diagnostic assays due to nonspecific detection or contamination; (b) larger volume of the samples (up to 100 times more) used for the tradition assays compared with that used for the multiplex PCR; and (c) atypical viruses or bacteria that have sequence substitutions at the primer or probe recognition sites.
Comparative performance of multiplex polymerase chain reaction (PCR) and traditional diagnostic assays in detecting Bovine coronavirus (BCoV), group A Bovine rotavirus (BRV), Salmonella spp., Escherichia coli K99+, or Cryptosporidium parvum from fecal samples.*
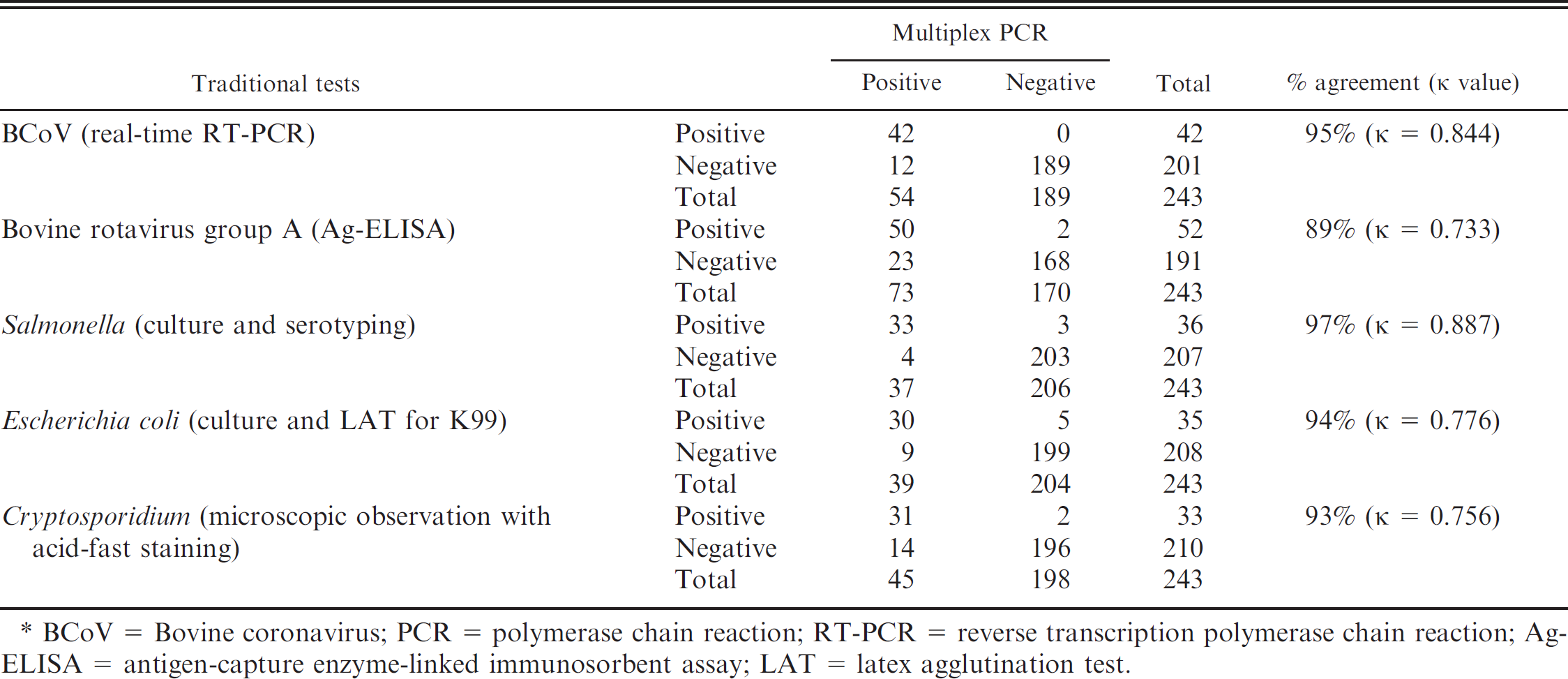
BCoV = Bovine coronavirus; PCR = polymerase chain reaction; RT-PCR = reverse transcription polymerase chain reaction; Ag-ELISA = antigen-capture enzyme-linked immunosorbent assay; LAT = latex agglutination test.
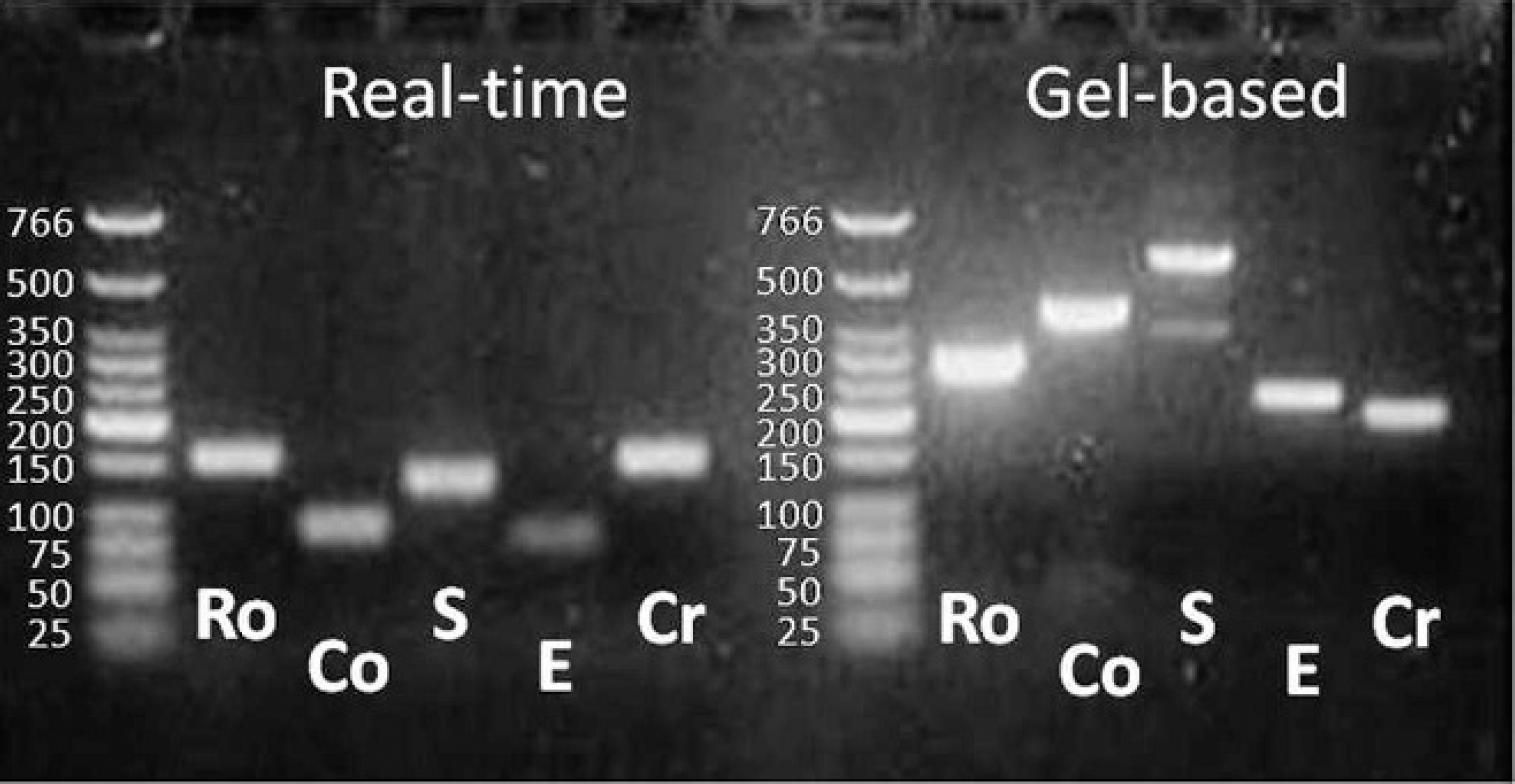
Electrophoretic analysis of polymerase chain reaction (PCR) products from group A Bovine rotavirus (Ro), Bovine coronavirus (Co), Salmonella sp. (S), Escherichia coli K99+ (E), and Cryptosporidium parvum (Cr) on 2% agarose gel. Extraction was made with a commercial nucleic acid isolation kit d PCR amplification was attempted with primers designed for multiplex real-time PCR and gel-based PCR as summarized in Tables 1 and 2, respectively.
Bovine coronavirus real-time PCR employed in the multiplex PCR panel has been developed based on a nested PCR described previously 7 and is being routinely used to detect BCoV from calf diarrheic cases at ISU VDL. The test results of the real-time PCR have shown good agreement with those of the nested PCR and correlated well with pathologic evidence and clinical history. The primers and probe for group A BRV were designed in the present study because no real-time RT-PCR that can detect a broad range of genetically diverse group A BRV has been reported previously. Inclusion of 3 pairs of forward and reversed primers and 2 probes, which were designed using the VP6 gene of BRV, was necessary to cover all known group A BRV based on the sequence database in GenBank, since the sequence homology of even VP6, which is highly conserved gene of BRV, ranged from 86% to 96% among group A BRV. The multiplex PCR appeared to detect most, if not all, of field BRV strains because it detected BRV virus in the 50 field samples that were also positive by a group A rotavirus Ag-ELISA kit, which is known to detect all group A rotavirus of various animal species including humans. However, both the multiplex and gel-based rotavirus PCR tests could not detect BRV in 2 samples that were positive by the ELISA. Presuming that the result of Ag-ELISA on those 2 samples were correct, it emphasizes the need for periodical updating of primers and/or probes to ensure that the PCR detects all circulating field strains, as RNA viruses are known to evolve rapidly. Another important point to carefully consider when interpreting the PCR results for BCoV and BRV is vaccination history, as oral vaccination with attenuated live BCoV and BRV vaccines are sometimes applied to newborn calves even though cow vaccination seems to be more commonly practiced in many bovine herds. Using electron microscopy, both BCoV and BRV were detected in calf feces until 3 and 7 days, respectively, after experimental vaccination with a BCoV-BRV multivalent attenuated vaccine via oral route. 27 Therefore, there is a great chance that the multiplex PCR detects both wild-type and vaccine viruses if attenuated vaccines are applied to newborn calves and samples are collected within such a timeframe.
The Salmonella PCR employed in the current multiplex PCR panel was reported to detect Citrobacter amalonaticus as Salmonella false positively, whereas other Citrobacter spp. were not detected by the multiplex PCR. 21 Nonetheless, C. amalonaticus did not seem to cause a significant problem in cattle, because the 72 fecal samples collected from healthy cattle were negative for Salmonella by the multiplex PCR, and only Citrobacter freundii were isolated from 2 samples out of 243 clinical samples by bacterial culture. However, those isolates were negative for Salmonella by PCR, suggesting that C. amalonaticus is not commonly detected in bovine species.
In conclusion, the newly developed multiplex PCR panel described in the current study is specific and more sensitive than other traditional diagnostic methods and drastically decreases turnaround time, labor, and cost. Therefore, the multiplex PCR panel could help diagnosticians rapidly determine the causative agents for bovine diarrhea in the early stages of disease and help practitioners initiate appropriate treatments or interventions quickly.
Acknowledgements
The authors thank Dr. Subhashinie Kariyawasam at Pennsylvania State University (State College, Pennsylvania) for providing K99+ E. coli PCR protocol. Dr. Jeffrey Knittel at Boehringer-Ingelheim Vetmedica Inc. (St. Joseph, Missouri) is acknowledged for providing Cryptosporidium-challenged samples, which were very valuable to this study. The authors are also grateful to Drs. Vickie Cooper, Bruce Leuschen, Terry Engelken, and Leo Timms at Iowa State University for advice in test evaluation. The study was supported in part by funding from Iowa Calf Scour Fund.
Footnotes
a.
U.S. Department of Agriculture, National Veterinary Services Laboratories, Ames, IA.
b.
American Type Culture Collection, Manassas, VA.
c.
Waterborne Inc., New Orleans, LA.
d.
MagMAX™ Total Nucleic Acid Isolation Kit, AgPath-ID™ Multiplex RT-PCR kit, ABI 7500 Fast Real-Time PCR System, MagMAX™ Viral RNA Isolation kit; Applied Biosystems, Austin, TX.
e.
TissueLyser, QuantiTect™ probe RT-PCR kit, QIAquick® PCR purification kit, QIAGEN® One-Step RT-PCR kit; Qiagen Inc., Valencia, CA.
f.
VWR International, West Chester, PA.
g.
KingFisher® 96, Thermo Fisher Scientific Inc., Waltham, MA.
h.
Version 3.0, Applied Biosystems, Foster City, CA.
i.
Integrated DNA Technologies Inc., Coralville, IA.
j.
QuikChange® II site-directed mutagenesis kit, Agilent Technologies (Stratagene products), La Jolla, CA.
k.
Smart Cycler® II, Cepheid Inc., Sunnyvale, CA.
l.
Premier™ Rotaclone® kit, Meridian Bioscience Inc., Cincinnati, OH.
m.
Difco Laboratories Inc., Sparks, MD.
n.
Remel Inc., Lenexa, KS.
o.
Sensititre®, Trek Diagnostic Systems, Cleveland, OH.
p.
BD, Franklin Lakes, NJ; or Statens Serum Institute, Copenhagen, Denmark.
q.
Pilitest™, VMRD Inc., Pullman, WA.
r.
Newcomer Supply Inc., Middleton, WI.
s.
DNASTAR Inc., Madison, WI.