
Abstract
Since 2007, outbreaks of severe bloody diarrhea and hemorrhagic colitis have been reported in the United States and Canada. Though the primary causative agent of swine dysentery is Brachyspira hyodysenteriae, which is strongly hemolytic, the current report describes the isolation of a novel strongly hemolytic Brachyspira sp. This novel Brachyspira sp. was identified from clinical submissions at the Minnesota Veterinary Diagnostic Laboratory, and 40 of such isolates were obtained from 22 clinical submissions representing 5 states. Isolates were confirmed to be different from any known Brachyspira sp. on the basis of phylogenetic analysis of nucleotide sequences of nox and 16S ribosomal RNA (rRNA) genes. Phylogenetic analyses grouped all isolates into 2 clades (clades I and II), and grouping patterns were similar for both nox and 16S rRNA gene sequence analyses. Phenotypically, all isolates were indole and hippurate negative, and enzymatic profiling indicated 2 types of profiles, irrespective of the phylogenetic grouping, differing only in the production of β-glucosidase. The results suggest that a potentially virulent new species of Brachyspira sp., provisionally named “Brachyspira hampsonii ”, is circulating among swine herds in the United States.
Introduction
The genus Brachyspira contains several Gram-negative, oxygen-tolerant, anaerobic intestinal spirochete species that colonize the large intestine of a wide range of animals including swine. 8 While some Brachyspira species cause intestinal disease in pigs, others are typically considered to be apathogenic commensals. 2 Brachyspira hyodysenteriae, which is the primary etiologic agent of swine dysentery, appears to be the most virulent and historically the most significant Brachyspira species in pigs. 9 Brachyspira hyodysenteriae is a strongly hemolytic species that causes a severe mucohemorrhagic diarrhea in pigs with high morbidity and low-to-moderate mortality. 8 Brachyspira hyodysenteriae primarily affects pigs during the growing-finishing period and has been reported from most swine-rearing areas of the world. Swine dysentery causes a significantly negative economic impact on the swine industry worldwide because of decreased growth rate, poor feed conversion, mortality, cost of medication and treatment, and restriction of the movement of stock.8,10
While B. hyodysenteriae is strongly hemolytic, other known species of Brachyspira are weakly hemolytic and appear to cause either a milder colitis (B. pilosicoli and B. intermedia) or no apparent disease at all (B. murdochii and B. innocens). Provisional Brachyspira spp. from other sources such as “B. canis” (from dogs), “B. pulli” (from chickens), and “B. corvi” (from corvid birds) have also been reported.4,11,19 In 2007, a new species of atypical strongly hemolytic Brachyspira, “B. suanatina”, was isolated from pigs in northern Europe that was found to be genetically distinct from all known swine Brachyspira spp. but very similar to isolates derived from mallards. 17 These isolates were recovered from pigs with swine dysentery and, in a challenge study, appeared to produce clinical signs and macroscopic changes consistent with the disease. 17 The findings suggest that several unrecognized Brachyspira species play an important role in clinically relevant swine intestinal disease. However, some species appear to be less of a clinical concern, and other potentially clinically relevant species and/or strains may still require appropriate taxonomic classification and/or recognition.
In the current study, phenotypic and molecular characterization of a novel strongly hemolytic (NSH) Brachyspira sp., isolated from clinical cases of swine dysentery in the United States, is described. The isolates were found to be different from all known Brachyspira spp. on the basis of nox gene sequencing, 16S ribosomal RNA (rRNA) sequencing, and biochemical testing, indicating a new species of Brachyspira, provisionally designated as “Brachyspira hampsonii ”. This name was selected to honor Dr. David Hampson of Murdoch University for his seminal work on the genus Brachyspira.
Materials and methods
Bacterial isolates
Cases of swine dysentery and related enteric diseases are routinely submitted to the Minnesota Veterinary Diagnostic Laboratory (MVDL; St. Paul, Minnesota) for disease investigation, and all field Brachyspira spp. used in the current study were obtained from the MVDL. For the isolation of Brachyspira spp. at MVDL, samples were plated onto BJ and CVS agar plates followed by incubation at 37°C under anaerobic conditions.12,13 After 5 days of incubation, all bacterial colonies expressing growth characteristics typical of Brachyspira spp. were selected for further identification by polymerase chain reaction (PCR). Hemolytic characteristics of each colony (i.e., whether the colony was strongly hemolytic or weakly hemolytic) on tryptic soy agar containing 5% sheep blood were also recorded at this time. In addition to clinical isolates, several American Type Culture Collection (ATCC) a control strains of Brachyspira spp. were used for phylogenetic analyses (Table 1).
Phenotypic characterization of novel strongly hemolytic (NSH) Brachyspira sp., provisionally designated as “Brachyspira hampsonii”.*
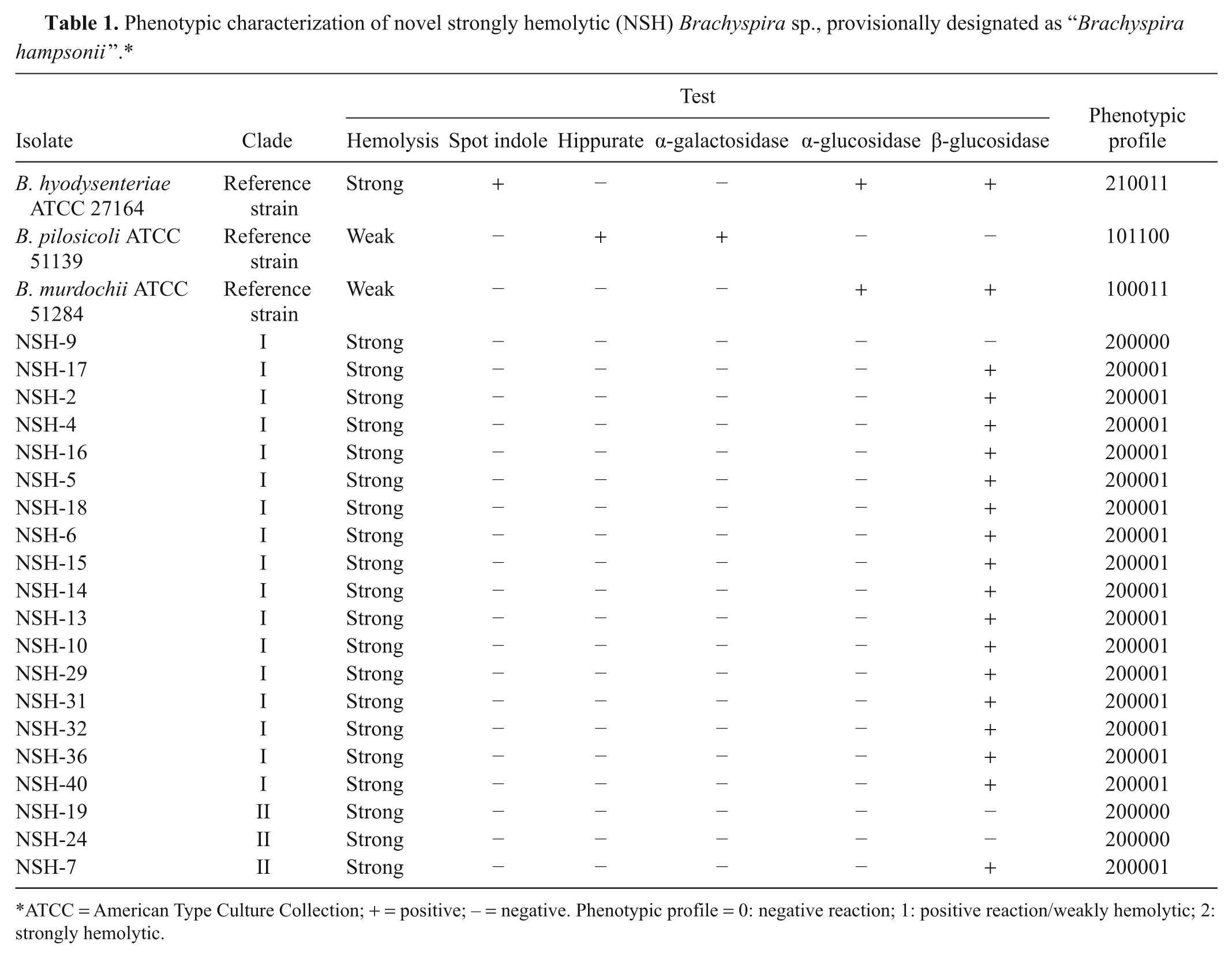
ATCC = American Type Culture Collection; + = positive; − = negative. Phenotypic profile = 0: negative reaction; 1: positive reaction/weakly hemolytic; 2: strongly hemolytic.
Brachyspira speciation
To identify Brachyspira isolates to the species level, DNA purified from each isolate was first tested using a previously described duplex PCR. 14 The duplex PCR method is capable of identifying Brachyspira isolates belonging to either B. hyodysenteriae or B. pilosicoli by targeting a 350-bp region of the 16S rRNA gene of B. pilosicoli and an 830-bp region of the NADH oxidase (nox) gene for B. hyodysenteriae.
All isolates that could not be identified using this species-specific duplex PCR (hereafter, “nontypeable” Brachyspira isolates) were identified by using a PCR and sequencing protocol, developed in the current study, which targets a highly variable region of the Brachyspira nox gene. 1 Primer designing software b was used to design the following primers: forward: 5′-GTTCTTGGCCTGTAACTCCTCCTAT-3′; reverse: 5′-GCAACAATACCCATTCTTACAG-3′. A 620-bp region of the nox gene was then amplified using these primers. After amplification, PCR products were purified using a commercial kit c and sequenced in both directions at the University of Minnesota Biomedical Genomics Center (St. Paul, MN) using an automated sequencer d with a commercial cycle sequencing kit e and the same primers as used for the PCR above. Sequences obtained were aligned using sequence alignment programsf,g to generate a sequence of approximately 550 bp, and this region of the nox gene was used for all subsequent phylogenetic analyses.
For further characterization of nontypeable Brachyspira isolates, the 16S rRNA gene was amplified by PCR using universal primers (forward: 5′-AGAGTTTGATCCTGG CTCAG-3′; reverse: 5′-ACGGCTACCTTGTTACGA CTT-3′). PCR products (approximately 1,480 bp) were purified using a commercial kit. c Purified products were sequenced in both directions using the same universal primers as used for the PCR. Sequences obtained were aligned to generate a sequence of approximately 1,380 bp, and this region of the gene was used for all subsequent phylogenetic analyses.
Phylogenetic analysis
For phylogenetic analyses, 1 isolate representing each clinical case (n = 22) was selected. Phylogenetic analyses of the nox and the 16S rRNA genes were performed separately. For nox gene analyses, nucleotide sequences for all the nontypeable Brachyspira isolates (n = 22), ATCC Brachyspira control strains, and additional Brachyspira isolate sequences from GenBank, were aligned using ClustalW and evolutionary distances were calculated by a neighbor-joining method. f For 16S rRNA gene analyses, nucleotide sequences for 18 out of 22 of the nontypeable Brachyspira isolates along with additional Brachyspira isolate sequences from GenBank, were aligned similarly as done for the nox gene. Full-length16S rRNA gene sequence of “Serpulina sp. P280/1” (not available in GenBank) was obtained from another laboratory. h
Multilocus sequencing typing
The 7 published multilocus sequencing typing (MLST) loci were amplified using the primers and PCR conditions as previously described.15,18 After PCR, purified PCR products were sequenced in both directions using the same universal primers as used for the PCR. Sequences were analyzed and assembled using a sequence analysis program. g The aligned sequences for each loci were matched with the online MLST database (www.pubmlst.org/brachyspira) to determine the number of alleles and identify sequence type according to allelic profile.
Phenotyping
Phenotypic characterization of isolates was done according to a previously developed system. 5 Biochemical and enzymatic reactions were performed as described below, and reaction patterns were indicated by a positive (+) or negative (–) reaction.
Beta-hemolysis. Isolates were cultured on sheep blood agar plates and were examined after 4–5 days of incubation at 37°C under anaerobic conditions. Beta-hemolysis was classified as weak or strong.
Indole production. The spot indole test was performed using James reagent. Briefly, growth from a culture plate was collected on a cotton swab and a drop of James reagent was added. A pink/red color confirmed a positive reaction, whereas no color indicated a negative reaction.
Hippurate hydrolysis. Test was performed using commercial discs i for detecting hydrolysis of sodium hippurate following manufacturer’s directions. A positive reaction was indicated by the development of a deep blue/purple color, whereas no color development indicated a negative reaction.
Enzymatic profile. Enzymatic profiles of isolates were determined using an enzyme-based strip identification system j following the manufacturer’s directions. After inoculation, the strips were incubated at 37°C for 5 hr, followed by the addition of 2 commercial reagents j to each well. Results were recorded as per the key provided in the kit. Positive (+) and negative (−) reactions were scored accordingly.
Results
Forty nontypeable Brachyspira sp. isolates were obtained from the MVDL during the period of 2009–2011. Each of the 40 isolates was derived from 1 of 22 different clinical cases of swine dysentery or related enteric disease that were submitted to the MVDL. All of the isolates were strongly hemolytic and tested negative (nontypeable) for B. hyodysenteriae and B. pilosicoli using a previously developed duplex PCR. 14 The isolates were subsequently evaluated using nox and 16S rRNA sequencing. Nucleotide analyses of obtained sequences using BLAST showed that these isolates most closely match an entry identified as “Serpulina sp. P280/1” (GenBank accession no. AF060815). The nox sequence from these nontypeable Brachyspira sp. isolates showed 95–96% homology with the “Serpulina sp. P280/1” nox gene. The next closest matches with respect to nox nucleotide sequence similarity are B. hyodysenteriae (92–93%), B. innocens (91–93%), B. murdochii (91–92%), and “B. suanatina” (91–92%). Each of the nontypeable Brachyspira isolates included in the analysis was strongly hemolytic. Because of this, and because these isolates do not clearly fall into any other Brachyspira sp. based on nox nucleotide sequence analysis, all 40 isolates were considered to be NSH Brachyspira sp. isolates.
Phylogenetic analysis of nox gene sequences grouped all NSH Brachyspira sp. isolates into 2 distinct monophyletic clades (I and II), each of which formed a cluster independent of other known Brachyspira spp. (Fig. 1). The majority of the isolates belonged to clade I (n = 29). Isolates within each NSH clade had a sequence similarity of >99%, whereas the sequence similarity between the 2 NSH clades was 96%. Nevertheless, NSH clade I isolates and NSH clade II isolates consistently clustered together using nox sequence phylogenetic analysis regardless of the parameters of the tree-building algorithm used. In addition, nox phylogenetic analysis indicates that the NSH Brachyspira sp. isolates are most closely related to a GenBank entry identified as “Serpulina sp. P280/1” (AF060815), 1 followed by the clade including B. murdochii and B. innocens (Fig. 1).
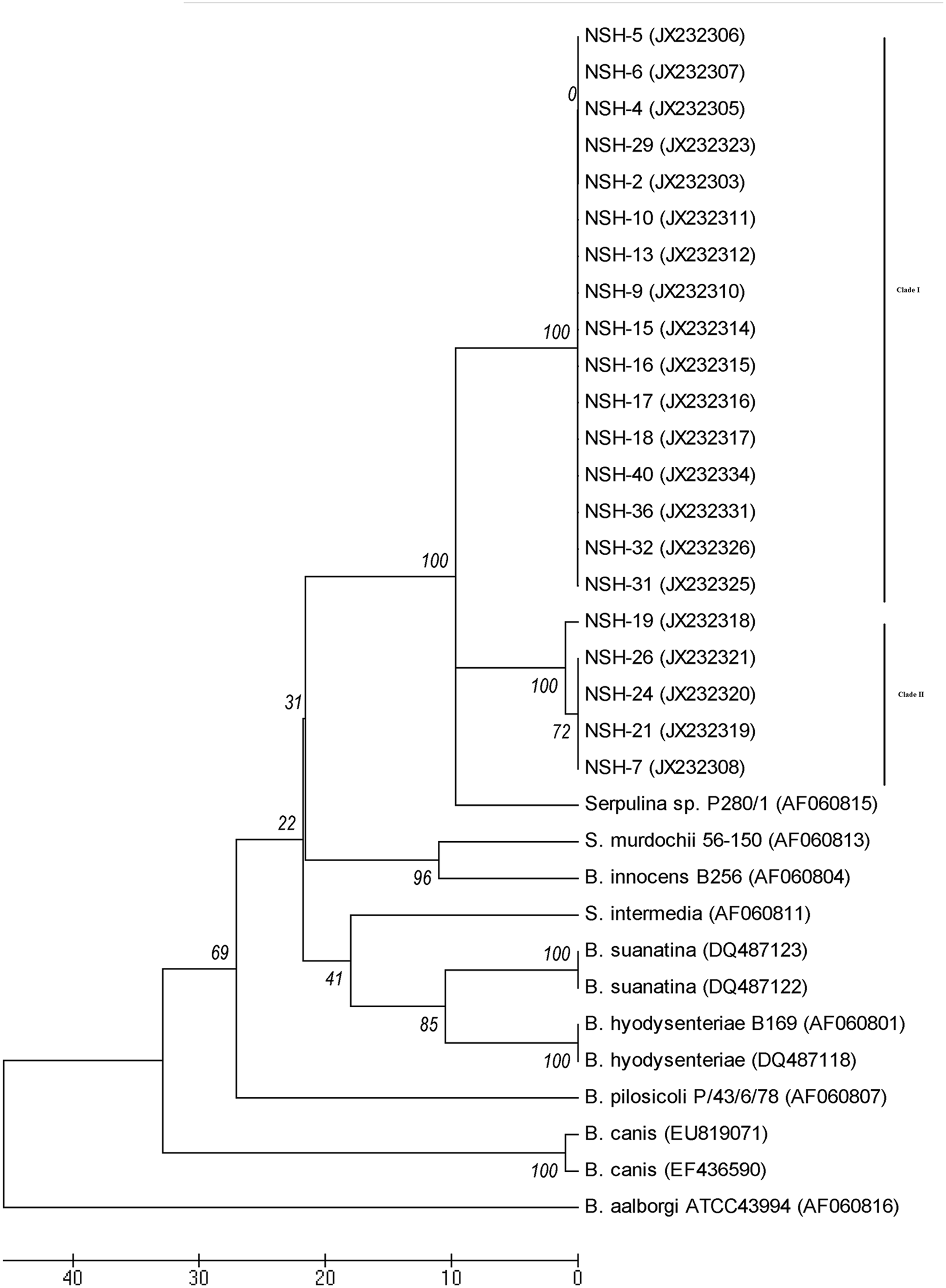
Phylogenetic analysis of the nox gene of Brachyspira sp. inferred using the neighbor-joining method and 100 bootstrap replicates (values shown on tree). The evolutionary distances were computed using the number of differences method, and evolutionary analyses were done in MEGA 5.f Sequences starting with NSH are from the novel strongly hemolytic (NSH) Brachyspira sp., provisionally designated “Brachyspira hampsonii ”, isolated in the present study, and the other sequences were obtained from GenBank and are identified with accession numbers.
A 16S rRNA gene sequence (approximately 1,380 bp) was obtained for NSH isolates from 18 of the 22 clinical submissions. Although the nucleotide sequence of this region of the 16S rRNA gene appears to be more conserved than the nox gene, the major findings of the 16S rRNA phylogenetic analyses are consistent with the nox analysis. All NSH Brachyspira sp. isolates tested fell into 2 monophyletic clades, the 2 clades consistently clustered near each other, and the isolates in these clades follow the same grouping pattern as for the nox gene (i.e., all isolates from NSH clade I based on nox analysis also clustered together in the 16S rRNA analysis, and all isolates from NSH clade II based on nox analysis also clustered together in the 16S rRNA analysis). Also, the NSH Brachyspira sp. isolates appear to be most closely related to “Serpulina sp. P280/1” and B. murdochii on the basis of 16S rRNA gene sequence (Fig. 2).
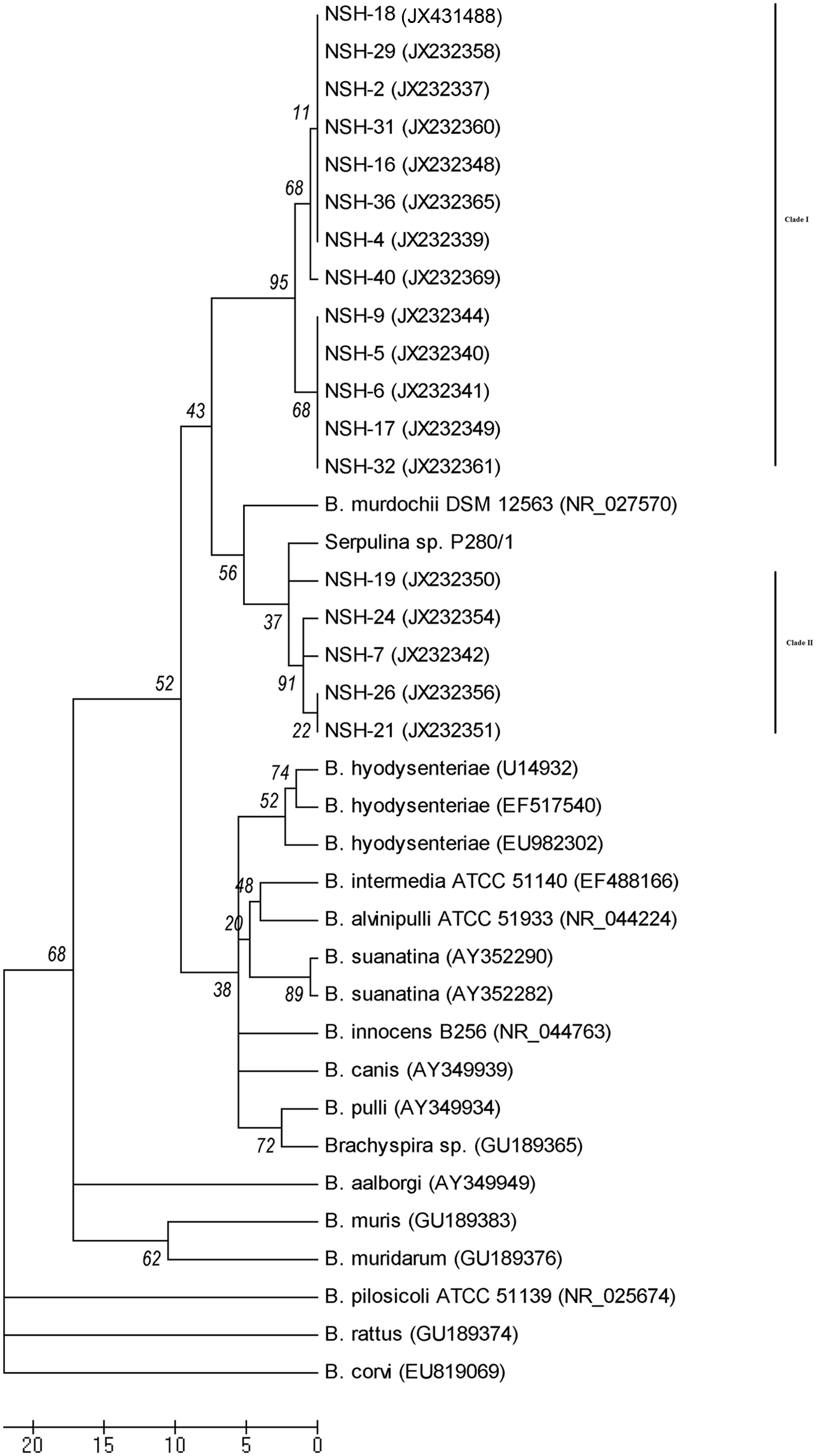
Phylogenetic analysis of 16S ribosomal RNA gene of Brachyspira sp. inferred using the neighbor-joining method and 100 bootstrap replicates (values shown on tree). The evolutionary distances were computed using the number of differences method, and evolutionary analyses were done in MEGA 5.f Sequences starting with NSH are from the novel strongly hemolytic (NSH) Brachyspira sp., provisionally designated “Brachyspira hampsonii”, isolated in the present study, and the other sequences were obtained from GenBank and are identified with accession numbers.
Of the 7 MLST loci, only 4 loci (est, pgm, glp, and thi) could be amplified for all NSH isolates. There was no amplification for the remaining 3 loci (adh, gdh, and alp) and, despite several attempts at optimizing PCR conditions, the 3 loci could not be amplified.
When sequences obtained for the 4 loci were matched with the MLST database, none of the loci gave exact matches with known alleles. The sequences for the thi gene gave the closest match with allele 24 of “Serpulina sp. P280/1” (Sequence Type-66), although there were some nucleotide differences (19 for clade I isolates and 26 for clade II isolates). For the other 3 loci (est, glp, and pgm), closest matches with alleles of Brachyspira sp. were observed and, while all isolates of clade II had similar allelic profile, isolates of clade I showed variation in their allelic profile (data not shown).
Phenotypic characterization results for 20 NSH Brachyspira sp. isolates (17 from NSH clade I and 3 from NSH clade II) are presented in Table 1. All NSH isolates that were tested were found to be indole and hippurate negative. Two different types of enzymatic profiles were observed; while all isolates were negative for α-glucosidase, most of the isolates tested positive for β-glucosidase. Although isolates from NSH clade I and NSH clade II could not be absolutely differentiated based on their enzymatic profiles, most of the isolates from NSH clade I were positive for β-glucosidase (16/17) and most isolates from NSH clade II were negative for this enzyme (2/3).
Discussion
Swine dysentery caused primarily by B. hyodysenteriae continues to be of global significance in grow-finish pigs. Identification of Brachyspira sp. is challenging. 17 Although PCR methods are useful for differentiation of B. hyodysenteriae and B. pilosicoli, PCR assays have been shown to have low sensitivities in detecting Brachyspira directly from clinical samples, 11 and perhaps more importantly, such assays are unable to identify many clinically relevant Brachyspira isolates to the species level.
Although swine dysentery has been largely absent from U.S. swine herds for the last 20 years, a number of recent outbreaks of severe bloody diarrhea and hemorrhagic colitis caused by Brachyspira pathogens have been reported in the United States and Canada. 3 In addition, the number of Brachyspira isolates recovered from clinical swine samples each year has been steadily increasing since 2002. 3 Furthermore, since 2008 more than 50% of the Brachyspira isolates recovered from pig intestines at the MVDL have been classified as “nontypeable” Brachyspira spp. as they could not be identified using current diagnostic PCR methods. Similarly, a 2011 report 3 stated that only 36% of the clinical isolates that were recovered at the Iowa State University Veterinary Diagnostic Laboratory in the first 9 months of 2010 could be identified to the species level using a conventional Brachyspira duplex PCR, further supporting the existence of a novel pathogenic Brachyspira currently in swine herds.
Each of the NSH Brachyspira sp. isolates that was characterized in the current study by sequencing the nox and 16S rRNA genes was cultured from a fecal or colon sample from swine facilities in 1 of 5 states (Iowa, Illinois, Minnesota, Missouri, and North Carolina). All of the isolates were found to be strongly hemolytic and were recovered from pigs presenting with enteric signs consistent with virulent Brachyspira infection, which suggests that the isolates are potentially virulent and associated with clinical disease in the field. Of the pigs from which samples were obtained, many were reported to have diarrhea, some with blood found in the stool. All of the isolates from these pigs tested negative for B. hyodysenteriae and B. pilosicoli during the course of their routine clinical diagnostic analysis using a diagnostic PCR.
On the basis of the nox gene sequence, all Brachyspira sp. isolates in the present study shared a homology of 83–93% with all known Brachyspira spp., whereas on the basis of the 16S rRNA gene sequence, all NSH Brachyspira sp. isolates showed homology of 96–99.4% with other known Brachyspira isolates. The disparity in sequence similarity between these 2 genes is not surprising, given the generally highly conserved nature of 16S rRNA genes and the high level of diversity that has been previously observed in Brachyspira nox genes. 1 The magnitude of divergence demonstrated at the nox locus for these NSH Brachyspira sp. isolates, however, taken together with the 16S rRNA sequence results and the phylogenetic analysis, strongly suggest that these NSH Brachyspira sp. isolates do not fall into any currently recognized species of Brachyspira.
Phylogenetic analysis of the obtained nucleotide sequences further indicated that the NSH isolates form 2 monophyletic clades (NSH clades I and II). This pattern was supported by both the nox and 16S rRNA phylogenetic analyses, although it is not perfectly clear from the analyses whether all of the NSH isolates (i.e., all NSH clade I isolates and all NSH clade II isolates) form a larger, single monophyletic clade or if the clade incorporating all of the NSH isolates studies should be considered paraphyletic. Isolates in clade II were found to be more closely related to “Serpulina sp. P280/1”, which formed a third subcluster in both the nox and 16S rRNA phylogenetic trees. “Serpulina sp. P280/1”, which was isolated from a pig with diarrhea in the United Kingdom, was also strongly hemolytic, indole negative, hippurate negative, negative for α-galactosidase and α-glucosidase, and positive for β-glucosidase in the commercial panel.j,16 Furthermore, “Serpulina sp. P280/1” was found to be pathogenic in gnotobiotic pigs. 16 It appears that isolates in NSH Brachyspira sp. clade II might have evolved from “Serpulina sp. P280/1”, whereas isolates in NSH Brachyspira sp. clade I evolved independently of clade II or “Serpulina sp. P280/1”; however, more studies are needed to verify this. In view of close relationship between all NSH Brachyspira sp. isolates in the current study and “Serpulina sp. P280/1”, the provisional name of “Brachyspira hampsonii” was proposed for this entire group of novel Brachyspira species, including clade I, clade II, and “Serpulina sp. P280/1”. If further discrimination were to indicate that these NSH Brachyspira isolates actually represent 2 separate species, the “B. hampsonii” species name would stay with clade II, with which “Serpulina sp. 280/1” is more closely related, whereas isolates in clade I will be renamed. The type strain for “B. hampsonii” clade I has been designated as isolate NSH-16, and the type strain for “B. hampsonii” clade II isolates has been designated NSH-24. Both of these type strains have been deposited with ATCC.FROM SOCIETY: Please provide ATCC numbers.
For the MLST analyses of the isolates in the present study, previously described primers and PCR conditions were used.15,18 Of the 7 MLST loci, only 4 could be amplified, and none of the amplified loci matched exactly with any of the alleles that are available in PUBMLST (www.pubmlst.org/brachyspira). Isolates from both groups had different MLST profiles for each of the 4 loci. These results further confirm that all of the NSH Brachyspira sp. isolates are different from any of the known Brachyspira species.
All NSH Brachyspira sp. isolates that underwent phenotypic characterization (n = 20) were found to be indole and hippurate negative, further distinguishing them, in general from B. hyodysenteriae (indole positive) and B. pilosicoli (hippurate positive). However, occasional indole-negative B. hyodysenteriae and hippurate-negative B. pilosicoli isolates have been reported.6,7 Using the commercial testing system, j 2 different enzymatic profiles were observed. The split of isolates based on the 2 different enzymatic profiles nearly matched the separation of NSH isolates based on nox and 16S rRNA sequence analyses, though there were a few exceptions; while most isolates from NSH clade I were positive for β-glucosidase (16/17), most isolates from NSH clade II were negative for this enzyme (2/3). Fewer isolates of clade II could be tested because some of isolates could not be revived from frozen stock cultures. Although one may expect to find a perfect association between phylogenetic differentiation based on genotype and enzymatic profile, others2,5 have also reported that isolates within the same group had more than 1 enzymatic profile.
Results presented in the current study indicate that a new and potentially virulent strain of strongly hemolytic Brachyspira is circulating among swine herds in the United States. Given the amount of genetic divergence between these novel isolates and other Brachyspira species, these NSH isolates likely represent a new species of Brachyspira, for which the provisional name of “Brachyspira hampsonii” is proposed. Although the determination as to whether these novel isolates represent a new species may seem like a semantic issue, the existence of significant genetic divergence between these isolates and other officially recognized Brachyspira species should not be overlooked, especially given the fact that these novel isolates are strongly hemolytic and phenotypically unique. The current findings highlight the importance of continued research on Brachyspira diversity, pathogenicity, and diagnostics.
Description of “Brachyspira hampsonii ”. “Brachyspira hampsonii” is a Gram-negative intestinal spirochete provisionally belonging to the genus Brachyspira. The species shares similar morphology and culture characteristics with other members of the genus. Members of the species can be distinguished genetically from other Brachyspira species by the sequence of their nox gene, their 16S rRNA gene sequence, and by the results of MLST analysis. “Brachyspira hampsonii” is strongly beta-hemolytic, but can be distinguished from the other strongly beta-hemolytic species (B. hyodysenteriae and “B. suanatina”) by being indole negative. Members of the species are negative for hippurate, alpha-galactosidase, and alpha-glucosidase. Strains of “B. hampsonii” clade I and clade II are generally beta-glucosidase positive and negative, respectively. The type strain for “B. hampsonii” clade I has been designated as isolate NSH-16, and the type strain for “B. hampsonii” clade II isolates has been designated NSH-24. Both of these type strains have been deposited with ATCC.
Footnotes
Acknowledgements
The authors would like to thank Dr. David Hampson and his laboratory (Murdoch University, Australia), for providing the 16S rRNA sequence of “Serpulina sp. P280/1”. The authors are also thankful to the Minnesota Veterinary Diagnostic Laboratory, College of Veterinary Medicine, University of Minnesota, for technical help.
a.
American Type Culture Collection, Manassas, VA.
c.
QIAquick PCR Purification Kit, Qiagen, Valencia, CA.
d.
ABI 373 XL DNA Sequencer, Applied Biosystems, Foster City, CA.
e.
BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems, Foster City, CA.
g.
Sequencher version 10.1, Gene Codes Corp., Ann Arbor, MI.
h.
Kindly provided by Dr. David Hampson, Murdoch University, Perth, Western Australia.
i.
BBL Taxo Hippurate discs, Becton, Dickinson and Company, Franklin Lakes, NJ.
j.
API ZYM, bioMérieux SA, l’Etoile, France.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.