
Abstract
A total of 30 nasal swabs from pigs preweaned and 11 nasal swabs from sick weaned pigs on a farm in Queensland, Australia, were cultured for the presence of Haemophilus parasuis. Enterobacterial repetitive intergenic consensus polymerase chain reaction (ERIC-PCR) genotyping and indirect hemagglutination and gel diffusion serotyping were performed on the retrieved H. parasuis isolates. A total of 3 genotypes were recognized among the 42 isolates recovered, and 4 representative isolates of each genotype were found to be nontypeable in the Kielstein/Rapp-Gabrielson serotyping scheme. A total of 20 of the 22 isolates of genotype 1 did not amplify in the species-specific conventional PCR number 1 (cPCR1) based on the 16S ribosomal RNA (rRNA) gene but did give the expected PCR amplicon in 2 other species-specific PCR assays, one of which is also based on the 16S rRNA gene. Nine selected isolates representing all genotypes, both positive and negative in the cPCR1, were sequenced, and all showed a 4-base mutation occurring at the forward primer annealing site. The quadruple base pair substitution from GTGG to TGTT near the 3' end of the forward primer sequence may explain the failure of amplification. Diagnostic laboratories should be aware that such failures can occur and should consider having an alternative PCR available to confirm negative results or, alternatively, use phenotypic characteristics for the identification of suspect H. parasuis isolates.
Isolates of Haemophilus parasuis, the causative agent of Glässer disease, can be identified by either of 2 conventional polymerase chain reaction (PCR) assays 2,13 or by a real-time PCR. 18 Problems with specificity have been encountered with conventional PCR 13 (hereafter referred to as cPCR1), because Actinobacillus indolicus gives a false-positive reaction. 13 An alternative conventional PCR (hereafter referred to as cPCR2) 2 has no reported problems with specificity but seems to be less sensitive than the cPCR1. 13 A recently developed real-time PCR 18 does not have known problems with specificity. When samples obtained from H. parasuis-challenged pigs were examined using the real-time PCR and cPCR1, the real-time PCR produced significantly more positive results. 18 There have been no reports in the veterinary literature of the failure of any of the 3 available PCR tests to recognize H. parasuis. The current work reports the failure of the cPCR1 13 to identify an H. parasuis strain and provides evidence that supports the reason for this failure.
Serovar profiling, performed as previously described, 17 was undertaken on a pig farm in Queensland, Australia. Samples were taken from the nasal cavities of 30 healthy, 3-week-old piglets (preweaning) from 15 sows ranging from parity 3 to 7 (2 from each sow). Nasal swabs were also collected from 11 pigs at approximately 8 weeks of age that were displaying clinical signs of illness, such as anorexia, swollen joints, and cyanosis. Two of these pigs had recovered from meningitis. Nasal swabs were cultured onto BA/SN agar 16 (blood agar base a supplemented with 0.0025% of NADH, 0.0005% of thiamine HCl, 1% of heat-inactivated horse serum, and 5% of oleic acid bovine albumin complex) 16 and, following overnight aerobic incubation at 37°C, single colonies characteristic for H. parasuis were subcultured onto BA/SN agar. Following overnight incubation, DNA was prepared by suspending a 1-μl loopful of bacteria in 100 μl of water, heating at 98°C for 5 min, cooling on ice for 5 min, centrifuging at 18,890 x g for 5 min, and retaining the supernatant at −220°C.
Isolates were confirmed as H. parasuis by the cPCR1. The DNA from isolates confirmed as H. parasuis and from isolates that were suspected to be H. parasuis were subjected to enterobacterial repetitive intergenic consensus (ERIC)-PCR, which was used to group the strains within the farm according to genotype profile. 13 Within each genotype, 4 representative isolates were serotyped by gel diffusion (GD) testing and indirect hemagglutination (IHA). 9,15 Isolates that did not react in any of the serological tests were regarded as nontypeable.
Fresh DNA template was produced as described above from the colonies growing on BA/SN plates for all isolates that failed to amplify in the cPCR1 plus 3 isolates that did amplify. The PCR was performed again using the new template. The PCR was also repeated twice more with the original template, which had been stored frozen at −20°C, for all isolates yielding positive and negative results in order to establish that these results were repeatable. A subset of 19 isolates were stored at −80°C and revived at a later stage. Template DNA was produced from the revived bacteria and tested again. A selection of 9 isolates were tested with the cPCR2 2 and with the real-time PCR. 18 All 19 stored isolates were processed with a modified forward primer (5'-GTGATGAGGAAGGTGTTTGT-3') in the cPCR1.
Details of results of all 3 polymerase chain reaction (PCR) assays (2 conventional PCR [cPCR] assays and 1 real-time PCR assay) on sequenced isolates of Haemophilus parasuis. *
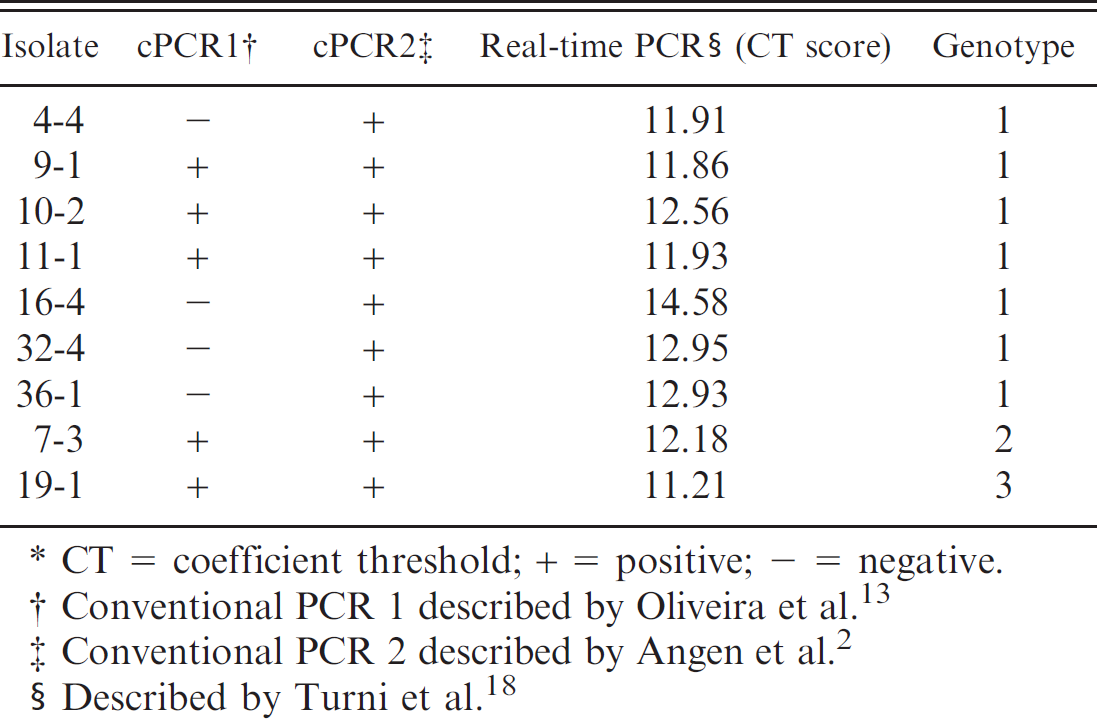
CT = coefficient threshold; + = positive; − = negative.
Conventional PCR 1 described by Oliveira et al. 13
Conventional PCR 2 described by Angen et al. 2
Described by Turni et al. 18
A conventional PCR targeting the 16S ribosomal RNA (rRNA) gene with universal bacteria primers 27F and 1525R (5'-GAGTTTGATCCTGGCTCAG-3' and 5'-AAGGAGGTGWTCCARCC-3', respectively) was run as previously described 3 with slight modification. Instead of a heating step before the addition of Taq DNA polymerase, the polymerase was added into the premix at the start. Another alteration was that the premix was briefly ultraviolet irradiated before the primers, and template was added to inactivate any contaminating DNA. The resulting PCR product was purified using a PCR purification kit b as per the manufacturer's instruction. A 4-μl volume of the purified PCR product from each isolate was sent to the University of Queensland Australian Genome Research Facility for sequencing together with 3 μl (3.2 pmol/μl) of each of the following primers: 27F, 519R, 530F, 787R, 926F, and 1525R. 10
The resultant sequences were aligned using the program Sequencher, c and a consensus sequence was produced for each isolate. The consensus sequence was compared to the reference strains for serovars 1, 4, and 5 and the type strain of H. parasuis (GenBank accession nos. FJ667948.1, FJ667951.1, FJ667952.1, and AY362909.1, respectively) using the Basic Local Alignment Search Tool (www.ncbi.nih.nlm.gov). 1
Full sequencing of the 16S rRNA gene was performed for the 9 isolates that were also investigated with the cPCR2 and the real-time PCR. Partial sequences of the 16S rRNA gene were obtained for the 19 stored samples using 3 primers (27F, 519R, and 787R). All 16S rRNA gene sequences were aligned with the sequence of the forward primer from the cPCR1.
From the nasal cavity of all pigs, with the exception of 1 sick pig, suspect H. parasuis isolates were recovered. These isolates demonstrated typical Haemophilus morphology; bacteria were satellitic around Staphylococcus hyicus on sheep blood agar, with a small colony size and an opaque, iridescent appearance on BA/SN 16 agar. A total of 22 samples were then identified by the cPCR1, and these isolates, plus an additional 20 more isolates that were PCR negative but which were suspected to be H. parasuis, were genotyped with an ERIC-PCR. 12 Three genotypes were identified. Serotyping revealed that all 3 genotypes were nontypeable in both the GD and IHA methods. In the 30 healthy pigs, 15 were colonized by genotype 1, 11 by genotype 2, and 4 by genotype 3. All genotypes were represented in the 11 sick pigs (7 sick pigs yielding genotype 1, 2 yielding genotype 2, 3 yielding genotype 3, and 1 that did not yield H. parasuis). All isolates pertaining to genotypes 2 (11 isolates) and 3 (7 isolates) tested positive using the cPCR1. However, for genotype 1, only 2 isolates gave a positive result, whereas 20 isolates yielded a negative result with the cPCR1. When new DNA template was prepared and retested, the 20 isolates still yielded a negative result. Repeating the cPCR1 twice more on all isolates resulted in the same negative and positive results as before. Following revival and preparation of fresh DNA template, the 19 stored isolates produced the same results (positive or negative) as were originally recorded.
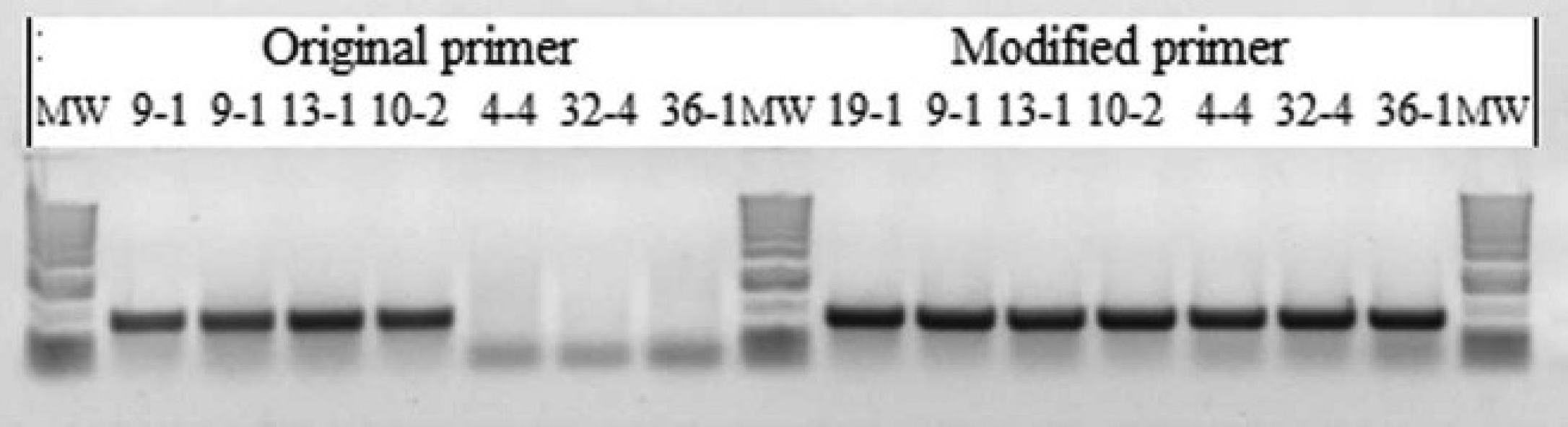
Results of the conventional polymerase chain reaction 1 14 using the original primers and with a modified forward primer, including the 4-bp substitution described in the text. MW = molecular weight marker lane.
A total of 3 isolates of genotype 1 and 1 isolate each of genotypes 2 and 3, which were all positive using the cPCR1, and 4 isolates of genotype 1, which gave a negative result using the cPCR1, were tested with the cPCR2 2 and the real-time PCR. 18 All isolates gave positive results using the cPCR2 and real-time PCR tests (Table 1). All 19 stored isolates yielded a positive result when tested with the cPCR1 13 with the modified forward primer (Fig. 1).
Nine isolates (Table 1) were subjected to full 16S rRNA gene sequencing, and all showed a 4-base mutation where the forward primer of cPCR1 binds (Fig. 2). This mutation is a quadruple-base pair substitution from GTGG to TGTT (corresponding to 446–449 bp of Escherichia coli American Type Culture Collection [ATCC] 11775T [accession no. X80725.1] and 428–431 bp of H. parasuis CCUG 3712T [accession no. AY362909.1]). There is also a quadruple-base pair substitution from TTAC to AGCA 16 bp further downstream in all 9 sequences (Fig. 2).
Targeted sequencing of the 16S rRNA gene region from the remaining 10 stored isolates showed that all stored isolates had the same 4-base mutations. The original target sequence of the forward primer for the cPCR1 was not found. None of the 9 isolates that were fully sequenced showed any change in the sequence of the cPCR1 reverse primer binding site.
There have not been any reports of failure of the cPCR1 to identify H. parasuis. The identification of 4 of the isolates that were negative in cPCR1 by the cPCR2 and real-time PCR tests as H. parasuis suggests that the failure lies within the primer region of the cPCR1. Mutation occurring at the primer annealing site resulting in loss or gain of a band or the change in its intensity has been reported with other PCR-based diagnostic tests. A single-nucleotide polymorphism at the 3'-end primer region causing mismatching and a failure of amplification has been reported for the molecular diagnosis of craniosynostosis syndrome, where 1 or more bones of the skull and face fuse prematurely during fetal development. 19 A mutation at the most 3' base of the initial reverse amelogenin gene PCR primer has been reported to interfere with gender identification in human beings. 14 Running the cPCR1 with the modified primer resulted in amplification, which suggests that the transversion did not result in stem-loop interference at the primer site. This would suggest that the quadruple-base pair substitution from GTGG to TGTT towards the 3' end of the forward primer found in the current study prevented the annealing of the primer, and therefore no amplification could occur.
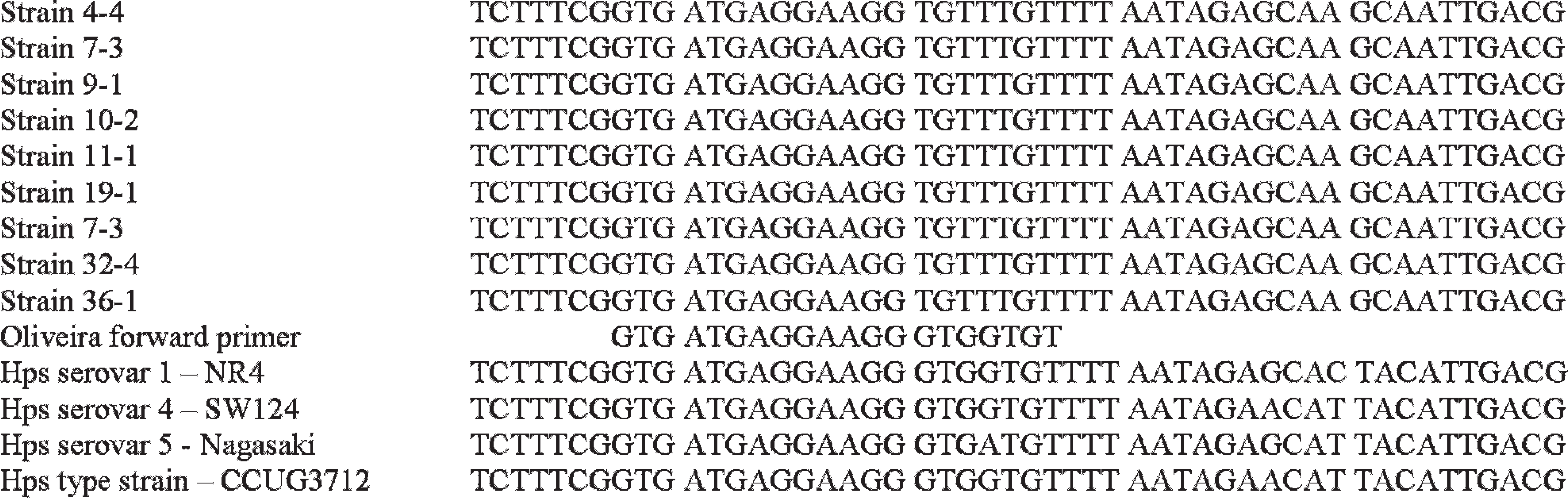
Sequence change at the conventional polymerase chain reaction 1 13 forward primer site and 16 bp downstream from the primer site.
Most bacteria have more than 1 copy of rRNA gene operons, 8 with the recently sequenced H. parasuis serovar 5 strain having 6 copies of the 16S rRNA gene.21 Studies have found that the amount of 16S rRNA amplification depended on 2 parameters: the genome size and the number of 16S rRNA genes. 5,6 The transversion described in the current study could mean the occurrence of the mutations in several copies of the 16S rRNA gene in all bacteria, or the occurrence of 2 subpopulations that are different at the relevant locus (wild type vs. mutant). 11 The fact that all sequenced isolates (i.e., isolates that were both negative and positive in the cPCR1) contained the substitution and the fact that all 19 stored isolates were positive with the modified primer suggest that each of the initially positive isolates has both copies of the gene (wild type and mutant), but the negative isolates only have the substituted version of the gene. The explanation that there are 2 subpopulations (wild type and mutant) does not seem plausible, because those strains that were consistently positive in the cPCR1 also all had the mutation. The fact that all of the positive isolates were consistently positive and were originally sampled from a single colony makes the subpopulation a less likely explanation. It would suggest that the isolates that gave a positive result with the cPCR1 still have the sequence recognized by the forward primer of the cPCR1, and therefore not all 16S rRNA genes have the transversion in these isolates.
Intragenomic heterogeneity between multiple 16S rRNA operons in sequenced bacterial genomes has been reported in many bacterial species. 4,7 The intragenomic heterogeneity between multiple 16S rRNA operons in sequenced bacterial genomes is rather limited, and transitions were the dominant type of mutation. 4 Most heterogeneity occurred in variable regions V1, V2, and, to a lesser extent, V3 + V4. 4 The mutation found in the current study was in the V3 region, even though it was of the less dominant type of mutation. The fact that it was in a variable region and that genomic heterogeneity is quite common in bacterial species makes the explanation of heterogenic copies of the 16S a very plausible explanation for the lack of amplification of some isolates.
The present report is the first recognition of false-negative reactions in any of the 3 recognized PCR tests for H. parasuis. Diagnostic laboratories should be aware of this possibility of false-negative reactions. Alternatives such as confirmation of negative results using other existing PCR tests or the use of phenotypic identification should be considered.
Acknowledgements
The authors would like to thank M. Pyke for his skilled technical assistance.
Footnotes
a.
BBL™, Becton Dickinson, Sparks, MD.
b.
QIAquick®, Qiagen GmBH, Hilden, Germany.
c.
Gene Codes Corp., Ann Arbor, MI.