
Abstract
A TaqMan real-time polymerase chain reaction (PCR) and loop-mediated isothermal amplification (LAMP) assay were developed to detect Gallid herpesvirus 1 (GaHV-1, formerly Infectious laryngotracheitis virus). The standard curve of real-time PCR was established, and the sensitivity reached 10 copies/μl. In the current study, the conversion between viral titer and GaHV-1 genomic copy number was constructed. Six primers for LAMP assay amplified target gene at 65°C within 45 min, and the detection limit was 60 copies/μl. The 6 primers were highly specific, sensitive, and reproducible for detection of GaHV-1. Although the sensitivity of LAMP was lower than that of real-time PCR, LAMP was faster, less expensive, and did not require a thermocycler. The LAMP assay would be a viable alternative assay in diagnostic laboratories that do not employ real-time PCR technology.
Keywords
Gallid herpesvirus 1 (GaHV-1; order Herpesvirales, family Herpesviridae, subfamily Alphaherpesvirinae, genus Iltovirus7,9,11,17; formerly Infectious laryngotracheitis virus) causes infectious laryngotracheitis (ILT), an acute respiratory disease of chickens, which occurs worldwide and causes economic losses. Procedures for ILT diagnosis include clinical signs, gross and histopathological lesions, and polymerase chain reaction (PCR).4,12,19 Real-time PCR is more sensitive than traditional methods, such as histopathological examination, cell culture isolation, direct fluorescent antibody test, and electron microscopy.3-5
The loop-mediated isothermal amplification (LAMP) assay is a DNA amplification method, which can be performed at a constant temperature of 60–65ºC in 60 min with Bacillus stearothermophilus (Bst) DNA polymerase. The LAMP uses 4 or 6 primers that recognize 6 or 8 regions on the target DNA sequence. When the reaction is initiated, the forward inner primer (FIP) attaches to the target DNA and synthesizes a complementary strand. The forward outer primer then anneals to the target DNA and initiates strand displacement DNA synthesis. The DNA strand, which is elongated from FIP, is replaced and released. The released single-stranded DNA forms a loop structure at its 3’ end. This released single-stranded DNA serves as a template for backward inner primer (BIP). The BIP attachment and DNA elongation is followed by a new BIP-primed DNA displacement using a backward outer primer–primed DNA, which leads to a “dumb-bell” form of DNA. This dumb-bell form DNA uses self-structure as the template. The elongation starts from FIP and BIP annealing to the single strand of the stem loop. After several runs of strand displacement and DNA synthesis, a mixture of stem-loop DNA with different length and cauliflower-like structures with several loops formed by annealing between alternately inverted repeats of the target within the same strand was produced.8,10,13,14,18
The present study reports the development and comparison of a LAMP assay with a novel real-time PCR for GaHV-1 DNA detection. Five ILT vaccines were used: AviPro LT, a LT BLEN, b Laryngo-Vac, c Trachivax, d and IT-IVAX. d Evaluation of specificity was conducted using the following non–GaHV-1 vaccines as controls: Mycoplasma gallisepticum vaccine, e fowl pox vaccine, c and Marek’s disease serotype 3 vaccine. c Two PCR-positive GaHV-1 field samples were also used.
Total DNA from vaccines was extracted, f according to the manufacturer’s instructions. To construct a plasmid for establishing the standard curve of real-time PCR and checking the sensitivities of both assays, the LT BLEN GaHV-1 partial infected cell protein 4 (ICP4) gene was amplified by PCR using the following primers: ICP4-F (5’-CGCAGAGGACCAGCAAAGACCG-3’) and ICP4-R (5’-GAAGCAGACGCCGCCGTAGGAT-3’). For PCR, the following reaction was performed in 50 µl: 5 µl of 10× PCR buffer, 5 µl of 25 mM MgCl2, 1 µl of 10 mM of each deoxyribonucleotide triphosphate, 1 µl of 10 µM ICP4-F, 1 µl of 10 µM ICP4-R, 0.25 µl of Taq DNA polymerase, g 28.75 µl of water, and 5 µl of sample DNA. The PCR steps were a 94°C initial denaturation for 2 min and 35 cycles of 94°C denaturation for 30 sec, 55°C annealing for 30 sec, and 72°C extension for 1 min, followed by 72°C final extension for 5 min. The PCR was conducted using a commercial system. g An expected 942-bp PCR product was detected in 2% agarose gel electrophoresis.
The PCR products were purified with a commercial purification system. h The complementary DNA was cloned into the pT7Blue-3 vector. A blunt-end cloning kit i was used according to the manufacturer’s instructions. Plasmids were transformed into NovaBlue Singles competent cells. i Several clones were selected, and the plasmid DNA was extracted using a commercial kit. h Clones containing the proper insert were verified by DNA sequencing. Concentrations of cloned plasmids were measured by a spectrophotometerj. Copy number of ILT ICP4-cloned DNA was calculated using the following formula:
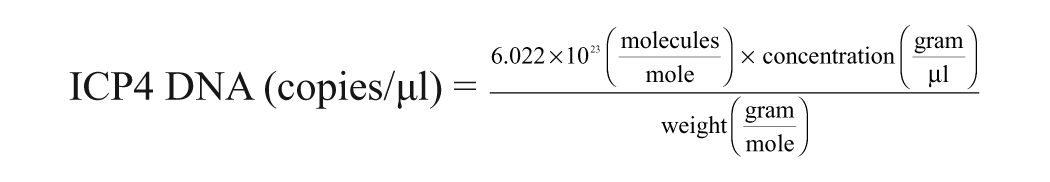
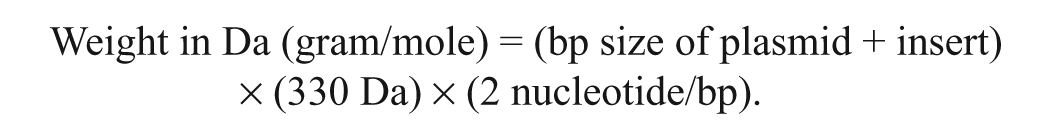
The real-time PCR amplification of a partial GaHV-1 ICP4 gene was performed in a 20-μl reaction volume using a commercial system. k Each reaction contained 10 µl of 2× master mix, f 1 µl of 10 µM each primer (0.5 µM), 0.5 µl of 4 µM probe (0.1 µM; Table 1), 2.5 µl of water, and 5 µl of DNA template. The real-time PCR program used 95°C activation for 15 min and 40 cycles for 95°C denaturation at 30 sec and a 60°C combined annealing and extension step for 60 sec.
TaqMan real-time polymerase chain reaction primers and probe sequences used in the current study.*

FAM = 6-carboxyfluorescein; TAMRA = 6-carboxytetramethylrhodamine.
The position numbers of the primers and probe were obtained from GenBank accession no. NC_006623.
The LAMP assay was performed in 25 µl, which contained 1× reaction buffer. l Each deoxyribonucleotide triphosphate was used at a final concentration of 1.2 mM, inner primers to a final concentration of 1.6 µM, outer primers to a concentration of 0.2 µM, loop primers to a concentration of 0.4 µM (Table 2), 1.0 M of betaine, m 1 µl of 8U Bst DNA polymerase, l 4 mM of MgSO4, and 5 µl of DNA template. For optimization of the reaction, 60°C, 63°C, and 65°C temperatures were tested. Reaction times of 15, 25, 35, 45, 50, and 60 min were examined for optimizing the LAMP assay. Reactions were stopped at 95°C for 3 min to terminate enzyme activity. After the LAMP reaction, DNA products were verified by 2% agarose gel electrophoresis, stained with ethidium bromide, and visualized on an ultraviolet transilluminator.
Primers for loop-medicated amplification assay used in the current study.

The primers and probe of the real-time PCR and LAMP assays were designed at the conserved region of the ICP4 gene and in the ICP4F/R amplicon. The partial ICP4 gene sequences of the following GaHV-1 strains were aligned from vector NTI sequence analysis and data management software version 10.3 n : CEO vaccine (GenBank accession no. EU104900), TCO vaccine (accession EU104908), the GaHV-1 assembled total genome sequence (GenBank NC_006623), 2 partial ICP4 gene sequences from field GaHV-1 strains (accession nos. L32139 and DQ 995291), and 2 GaHV-1 vaccines (AviPro LT and LT BLEN), which were sequenced at Auburn University Genomics & Sequencing Laboratory (Auburn, Alabama). Real-time PCR primers amplified a 125-bp product. The LAMP primers were designed using commercial software. o
To determine the sensitivity and detection limit for real-time PCR, serial 10-fold dilutions of plasmid DNA from 100–10 9 copies/µl were analyzed. The real-time PCR assay was repeated 4 times. Standard curves, indicating the linear relationships between the threshold crossing points (Cp) and the logarithms of initial GaHV-1 ICP4 gene count, were constructed. Serial dilutions were repeated in the same run to evaluate intra-experiment reproducibility. Serial dilutions of the ICP4 plasmids from 6, 15, 30, 60, 6 × 102, 6 × 103, and 6 × 104 copies/µl determined the sensitivity of the LAMP assay. Five GaHV-1 vaccine strains, 2 positive field strains, a non-template negative control, M. gallisepticum, fowl pox, and Marek’s disease vaccines were tested by real-time PCR and LAMP assays.
For investigating the conversion between viral titer and GaHV-1 DNA copy number, GaHV-1 vaccine stock was titrated in specific pathogen–free (SPF) chicken embryos, and the copy number of vaccine stock was determined with real-time PCR. The titration, 50% embryonic infective dosage (EID50) of the LT BLEN vaccine, was initiated using 9 serial 10-fold dilutions of viral stock prepared in phosphate buffered saline. Four 9-day-old SPF embryos were used per viral dilution, and each embryo was inoculated with 200 µl of viral dilution via the route. 16 After 7 days, chorioallantoic membrane routes were checked for GaHV-1–associated pock formation. Viral titer was calculated using the Reed–Muench formula. 15 The EID50 titrations were repeated 3 times, and the GaHV-1 genome copy number was detected with real-time PCR. Viral concentration was detected using 1 dose of LT BLEN vaccine. The vaccine virus was diluted using the manufacturer’s protocol to 4 doses, and the GaHV-1 genome copy number was checked by real-time PCR. The tests were repeated 3 times.
Both assays were positive for GaHV-1 DNA only. No fluorescent signal or positive gel patterns were detected with the non-template control and non–GaHV-1 vaccines. Four repeats of real-time PCR tests generated standard curves, with an average intercept of 38.28 ± 0.63 and an average slope of −3.14 ± 0.06. The standard curves had a significant correlation between Cp value and copy number with the square of the sample correlation coefficient (R 2 ) above 0.99 and an average efficiency was 2.063 ± 0.048. The standard deviation of Cp values at the same genomic copy number of multiple repeats was low, which indicated excellent reproducibility. The regression equations of standard curves maintained linearity at 10 copies/µl, and the standard deviation and standard error were stable and low. One of 4 replicated real-time PCR tests at 1 copy/µl was negative, and the Cp values of the other 3 repeats were above 35. Quantification limit of real-time PCR was determined using 10 copies/µl. Samples with Cp values ≤35 were considered positive for GaHV-1 DNA. The LAMP assay detected GaHV-1 DNA at 60 copies/µl. A SYBR Green I–based real-time PCR detected 140 gene copies/µl of GaHV-1 DNA. 2 It has been reported that a real-time PCR, using the GaHV-1 gC gene, detected 100 copies/reaction. 1 The sensitivity of current real-time PCR was higher than the LAMP and previous real-time PCR assays.
One EID50 of GaHV-1 vaccine was equal to (2.1 ± 1.3) × 10 2 GaHV-1 genomic copies, and 1 dose of GaHV-1 vaccine was equal to (6.9 ± 0.85) × 10 5 copies. The optimal temperature for Bst DNA polymerase was 65°C. The LAMP products were observed with bright bands after the LAMP reaction was performed more than 45 min. The electrophoresis photo of serial 10-fold dilutions of GaHV-1 ICP4-cloned DNA and GaHV-1–positive products showed many bands with different sizes and a smeared DNA between these bands.
Although the real-time PCR assay was more sensitive than the LAMP, the GaHV-1 LAMP did not require a thermocycler, expensive reagents, or molecular training of personnel. The LAMP took less than 1 hr compared to 2–4 hr for the real-time PCR. In addition, the sensitivity of LAMP assay was less affected by contaminating components, feces, feed, and blood, which can occur in clinical samples, than for real-time PCR.6,8,13,20 However, unlike conventional PCR or real-time PCR, the LAMP products cannot be sequenced directly, and the LAMP assay needs complicated primer sets. Five vaccine and 2 field GaHV-1 strains were tested in the current assays, but more field samples need to be checked for further validation before the test can used in diagnostic laboratories.
Footnotes
a.
Lohmann Animal Health Inc., Winslow, ME.
b.
Merial Select Inc., Gainesville, GA.
c.
Chicken-N-Pox™ TC, MD-Vac® CFL; Fort Dodge Animal Health, Overland Park, KS.
d.
Schering-Plough Animal Health Corp., Union, NJ.
e.
Mycovac-L®, Intervet Inc., Millsboro, DE.
f.
DNeasy® Blood & Tissue Kit, QuantiTect® Probe PCR kit; Qiagen Inc., Valencia, CA.
g.
GeneAmp® PCR System 9700, Applied Biosystems, Foster City, CA.
h.
Wizard® PCR preps DNA purification system, Wizard® Plus SV kit; Promega Corp., Madison, WI.
i.
Novagen, Merck KGaA, Darmstadt, Germany.
j.
NanoDrop™ ND-100UV-Vis spectrophotometer, Thermo Fisher Scientific, Wilmington, DE.
k.
LightCycler®, Roche Diagnostics Corp., Indianapolis, IN.
l.
ThermolPol, New England Biolabs Inc., Ipswich, MA.
m.
Sigma-Aldrich, St. Louis, MO.
n.
Invitrogen Corp., Carlsbad, CA.
o.
Primer Explorer V4, Eiken Chemical Co. Ltd, Tokyo, Japan.
The author(s) declared no conflicts of interest with respect to the research, authorship, or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.