
Abstract
Neuronal ceroid lipofuscinosis (NCL) constitutes a group of recessively inherited lysosomal storage diseases that primarily affect neuronal cells. Such diseases share certain clinical and pathologic features in human beings and animals. Neuronal ceroid lipofuscinosis in Border Collie dogs was first detected in Australia in the 1980s, and the pathogenic mutation was shown to be a nonsense mutation (c.619C>T) in exon 4 in canine CLN5 gene. In the present study, novel rapid genotyping assays including polymerase chain reaction (PCR)–restriction fragment length polymorphism, PCR primer–induced restriction analysis, mutagenically separated PCR, and real-time PCR with TaqMan minor groove binder probes, were developed. The utility of microchip electrophoresis was also evaluated. Furthermore, a genotyping survey was carried out in a population of Border Collies in Japan using these assays to determine the current allele frequency in Japan, providing information to control and prevent this disease in the next stage. All assays developed in the current study are available to discriminate these genotypes, and microchip electrophoresis showed a timesaving advantage over agarose gel electrophoresis. Of all assays, real-time PCR was the most suitable for large-scale examination because of its high throughput. The genotyping survey demonstrated that the carrier frequency was 8.1%. This finding suggested that the mutant allele frequency of NCL in Border Collies is high enough in Japan that measures to control and prevent the disease would be warranted. The genotyping assays developed in the present study could contribute to the prevention of NCL in Border Collies.
Keywords
Introduction
Neuronal ceroid lipofuscinosis (NCL) constitutes a group of recessively inherited lysosomal storage diseases that primarily affect neuronal cells. Such diseases share certain clinical and pathologic features in human beings and animals. 10 Typical clinical signs of these progressive neurodegenerative diseases include behavioral abnormality, sleep problems, mental retardation, dementia, seizure, motor abnormality such as ataxia, and/or in most cases visual problems leading to blindness.10,11 In veterinary medicine, NCL has been described in several domestic species and occurs most commonly in the dog. 12
Neuronal ceroid lipofuscinosis is characterized by massive accumulations of autofluorescent lysosomal storage bodies in the lysosomes of a variety of cell types, particularly neurons and retinal cells. 10 Accumulation of subunit c of mitochondrial adenosine triphosphate synthase has been demonstrated as the dominant storage protein in most human forms of NCL, as well as in cattle, horses, and several breeds of sheep and dog.11,29 In contrast, sphingolipid activator proteins A and D are the major protein components of storage bodies in a few human forms of NCL,19,27 in affected Swedish Landrace sheep, 28 Miniature Schnauzer dogs, 20 and Polish Lowland Sheepdogs. 18 However, the mechanisms of storage and neurodegeneration in NCL remain to be explained.
In canine NCL, mutations in several genes responsible for the disease were reported in English Setters (CLN8), 15 Border Collies (CLN5), 17 Bulldogs (CTSD), 3 Miniature longhaired Dachshunds (TPP1), 2 Miniature Dachshunds (PPT1), 23 American Staffordshire Terriers (ARSG), 1 Australian Shepherds (CLN6), 14 and Tibetan Terriers (ATP13A2). 7 Thus, once a mutation is identified in a breed or family, rather simple procedures can be used for a rapid genetic diagnosis and genotype screening leading to control and eradication of the disease.
Neuronal ceroid lipofuscinosis in Border Collies was first detected in Australia in the 1980s.8,24-26 A diagnosis of the first case in Japan was made in 2002. 16 In 2005, the pathogenic mutation was demonstrated to be a nonsense mutation (c.619C>T) in exon 4 in the canine CLN5 gene, 17 which enabled the development of rapid and simple genetic tests. Therefore, allele frequency should be determined accurately using these newly developed assays to estimate the requirements for preventing this disease in the next stage.
Polymerase chain reaction (PCR)-based genetic assays are very important for the prevention of a single gene disorder because these assays can detect not only affected individuals but also heterozygous carriers. In the original report regarding the pathogenic mutation of NCL in Border Collies, a PCR–restriction fragment length polymorphism (RFLP) assay was shown as a genotyping test (conventional PCR-RFLP assay), but it seems difficult to judge the genotypes because of complex fragmentation after digestion by restriction endonuclease. 17 Therefore, the present study developed and evaluated novel rapid and simple genotyping assays including improved PCR-RFLP, PCR primer–induced restriction analysis (PIRA), mutagenically separated (MS)-PCR, and real-time PCR with TaqMan minor groove binder (MGB) probes for NCL in Border Collies. The utility of the microchip electrophoresis was also evaluated in each PCR-based assay. Furthermore, genotype screening was carried out in a population of Border Collies in Japan using these assays to determine the current allele frequency in Japan and provide information to control and prevent this disease in the next stage.
Materials and methods
Samples and treatment
In the present study, control samples for each genotype were obtained from an affected Border Collie that had been diagnosed by histopathologic examination, a heterozygous carrier that was the sire of the affected dog and an unaffected Beagle. The genotypes were confirmed using direct sequence analysis. DNA templates for each genetic test were prepared using whole blood spotted onto Flinders Technology Associates filter paper (FTA card) a or saliva spotted onto indicating FTA card, a which had been stored at approximately 4ºC until used. For PCR-RFLP, PCR-PIRA, and MS-PCR, a 1.2 mm in diameter disc punched out of the FTA card using a hole punch a was used for a DNA template after treatment as follows. The disc was placed into a 0.2-ml PCR tube. The disc on the tube bottom was washed three times for 5 min each with 100 μl of washing solution a at room temperature, rinsed twice for 5 min each with 200 μl of Tris–ethylenediamine tetra-acetic acid (EDTA) buffer (pH 8.0) b and dried at 60ºC for 10 min. The treated disc was used directly as a template in all PCR tests except real-time PCR.
Conventional and improved PCR-RFLP methods
Each PCR test was carried out targeting a sequence around the mutation (c.619C>T) in exon 4 of the canine CLN5 gene with primers shown in Table 1. The conventional PCR-RFLP assay reported previously 17 was carried out with forward (cRFLP-F) and reverse (cRFLP-R) primers in a 20-μl reaction mixture containing 10 μl of 2× PCR master mix, c 12.5 pmol of primers, and the treated disc of FTA card as a template. After the first denaturation at 95ºC for 12 min, 40 cycles of amplification were carried out, at a denaturing temperature of 95ºC for 30 sec, an annealing temperature of 59ºC for 1 min, and an extension temperature of 72ºC for 1 min. Extension during the last cycle was carried out at 72ºC for 6 min. The product of the conventional PCR-RFLP was digested with the restriction enzyme MseI at 37ºC for 90 min in a 10-μl reaction mixture containing 7 μl of the product, 10 U of MseI, d 1 μl of 10× restriction enzyme buffer, d and 0.1 μl of 100× bovine serum albumin d included by the manufacturer.
Characteristics of primers and TaqMan probes used in the present study.*
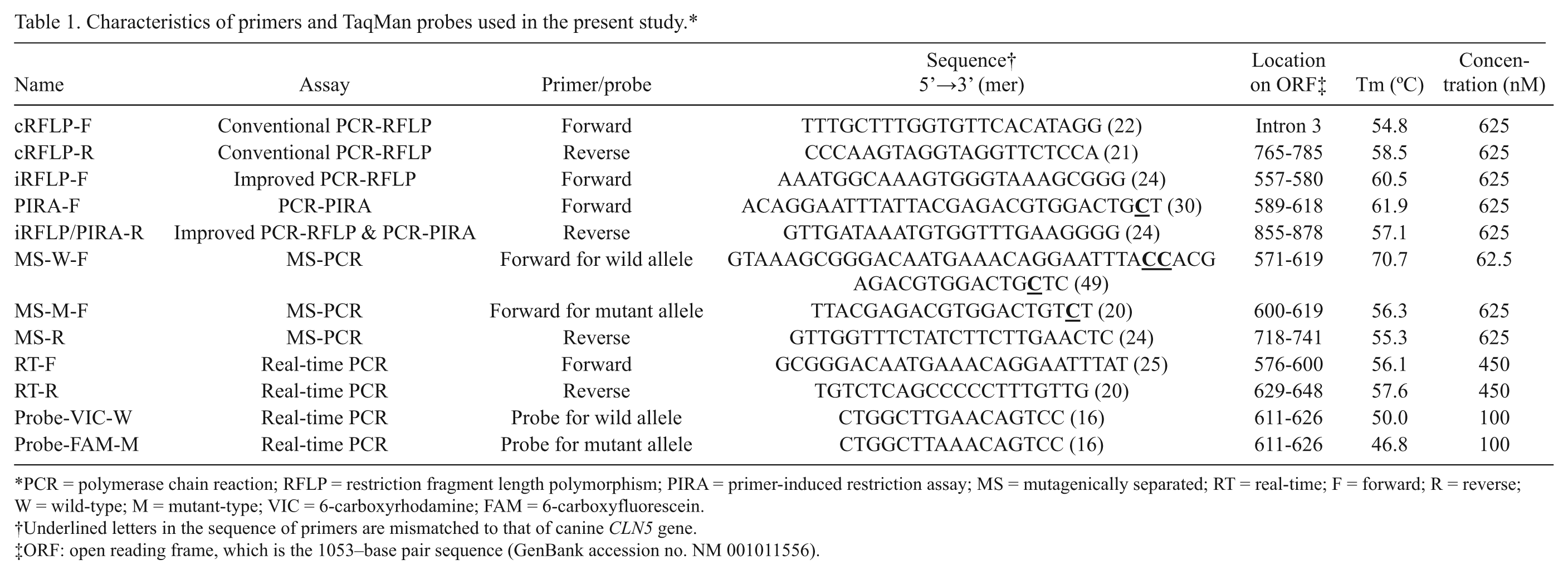
PCR = polymerase chain reaction; RFLP = restriction fragment length polymorphism; PIRA = primer-induced restriction assay; MS = mutagenically separated; RT = real-time; F = forward; R = reverse; W = wild-type; M = mutant-type; VIC = 6-carboxyrhodamine; FAM = 6-carboxyfluorescein.
Underlined letters in the sequence of primers are mismatched to that of canine CLN5 gene.
ORF: open reading frame, which is the 1053–base pair sequence (GenBank accession no. NM 001011556).
The improved PCR-RFLP was carried out using the same PCR reagents as those in the conventional PCR-RFLP with forward (iRFLP-F) and reverse (iRFLP/PIRA-R) primers that were designed to reduce the number of digested DNA fragments compared to that in the conventional PCR-RFLP method (Table 1). After the first denaturation at 95ºC for 2 min, 40 cycles of amplification were carried out, at a denaturing temperature of 95ºC for 30 sec, an annealing temperature of 55ºC for 30 sec, and an extension temperature of 72ºC for 1 min. Extension during the last cycle was carried out for 6 min. The condition of restriction enzyme digestion was the same as that in the conventional PCR-RFLP method.
PCR-PIRA method
The PCR-PIRA method can be used to detect a single nucleotide mutation by introducing an artificial restriction endonuclease site using primers containing mismatches. 9 The PCR-PIRA assay in the present study was carried out with forward (PIRA-F) and reverse (iRFLP/PIRA-R) primers. The forward primer introduced the restriction site (CT619TAAG) of restriction enzyme AflII on the mutant allele as a result of combination with the mutant genomic sequence (c.619T). The conditions of PCR and restriction enzyme digestion by AflII d (20 U) were the same as those in the improved PCR-RFLP method.
MS-PCR method
The MS-PCR method utilizes sequence-specific hybridization by 2 primers with widely different sizes and was originally developed for easier genotyping of human familial hypercholesteremia in a single PCR tube without a restriction endonuclease digestion process. 22 In the present study, the MS-PCR assay was designed based on the general rules of MS-PCR, 22 and the ratio of short to long allele-specific forward primers (MS-W-F and MS-M-F) was increased to 10:l on a molar basis (Table 1) in order to produce bands of similar intensity in the heterozygous carrier when visualized on gel electrophoresis. The PCR assay was carried out using these 2 allele-specific forward primers and a reverse primer (MS-R) simultaneously in a single PCR tube in which the reaction condition was the same as that in the abovementioned PCR methods except for the annealing temperature (50ºC).
Agarose gel and microchip electrophoreses
All undigested and digested PCR products except for the real-time PCR were subjected to electrophoresis in 3% (w/v) agarose gel e dissolved with Tris–EDTA buffer. b Molecular size markers f were used in the agarose gel electrophoresis. The electrophoresed agarose gel was stained with ethidium bromide, and luminescence was induced by an ultraviolet transilluminator. Analysis of PCR products was also performed using a microchip electrophoresis system g with a special reagent kit g that included internal DNA size markers and DNA separation buffer. A fluorescent dye h was added to the DNA separation buffer according to the manufacturer’s protocol for the microchip electrophoresis system. The PCR products were diluted 10 times with deionized water before application to this system to diminish the adverse influence of PCR and endonuclease buffers on electrophoretic mobility. DNA ladder markers h from 25 to 450 base pairs (bp) were used as references for DNA sizing.
Real-time PCR method
Using a hole punch, a 1.2-mm in diameter disc was punched from the FTA cards described above. a The disc was placed into a separate 0.2-ml tube, lysed in the tube with 8 μl of lysis solution from a special DNA extraction kit, i and subsequently incubated at 95ºC for 3 min. Then, 8 μl of DNA stabilizing solution from the kit i was added to the tube. This DNA-containing solution was transferred to a new tube and stored at −25ºC until analysis.
Amplifications were performed on a real-time PCR system i using a specific primer pair and TaqMan MGB probes bound with each fluorescent reporter dye (6-carboxyrhodamine or 6-carboxyfluorescein) at the 5’-end and a nonfluorescent quencher dye at the 3’-end (Table 1), which were synthesized by a commercial facility. i The real-time PCR amplifications were carried out in a final volume of 10 μl consisting of a master mix (2×), i a genotyping assay mix (80×) i including specific primers and TaqMan MGB probes, nuclease-free water, and 2 μl of the aforementioned DNA-containing solution as a DNA template. The holding stage before PCR was performed at 25ºC for 30 sec. The cycling conditions were 20 sec at 95ºC followed by 40 cycles of 3 sec at 95ºC and 20 sec at 60ºC. The holding stage after PCR was performed at 25ºC for 30 sec.
In addition, an allelic discrimination plot was constructed based on the 3 types of amplification plots. Those data were calculated using software i based on the results obtained using DNA samples from 46 Border Collies (19 noncarriers, 19 carriers, and 8 affected) in which the genotypes were determined by the other PCR tests developed in the present study.
Genotyping survey
The genotyping survey was carried out using DNA samples from saliva- or whole blood–spotted FTA cards of 407 Border Collies in Japan. The samples were randomly collected with owners’ informed consent for scientific evaluation of their dogs’ DNA. Sample collection was performed from 2006 to the present by the Japan Border Collie Health Network, a volunteer breeders’ association for the healthy breeding of Border Collies. The genotypes were determined using one of the assays developed in the present study. Since the real-time PCR method was established, genotyping has been carried out using the real-time PCR assay only. The results obtained by the PCR-RFLP, PCR-PIRA, and MS-PCR assays were confirmed again by real-time PCR assay.
Results
Conventional PCR-RFLP assay
In the conventional PCR-RFLP assay, a 290-bp DNA band was amplified in theory in all the genotypes (Fig. 1). Since the amplification product in the unaffected dog had 3 recognition sites of the restriction endonuclease MseI, it was cleaved into 4 fragments (i.e., 129-, 101-, 38-, and 16-bp bands) after digestion with MseI. In the affected dog, because there were 4 recognition sites of MseI due to the mutation, the amplified band was cleaved into 5 fragments (i.e., 101-, 65-, 62-, 38-, and 16-bp bands), which appeared as 4 fragments due to the inseparability of 65- and 62-bp bands even on microchip electrophoresis (Fig. 1B). The amplification product in the carrier was cleaved into 6 fragments, which appeared as 5 fragments (i.e., 129-, 101-, 65-, 62-, 38-, and 16-bp bands). The 16-bp band was too small to be seen clearly under the screen of primer bands.
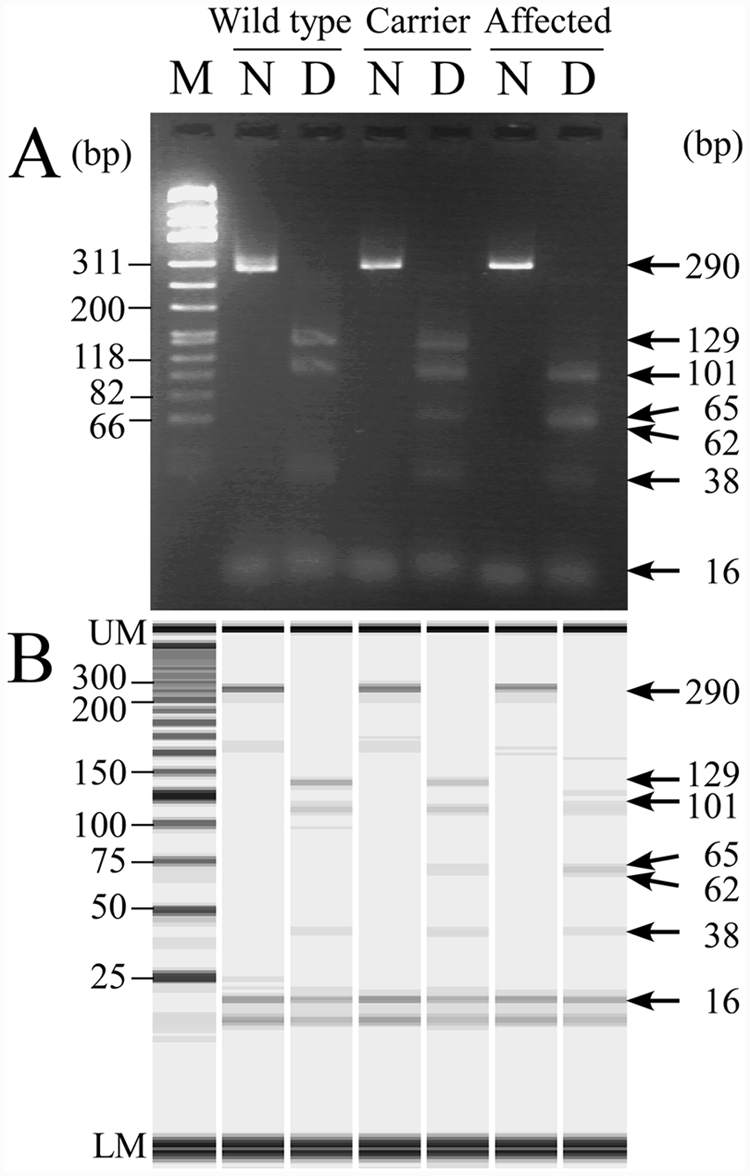
Neuronal ceroid lipofuscinosis genotyping of wild-type, heterozygous carrier and affected dogs by conventional polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) assay using agarose gel electrophoresis (
Improved PCR-RFLP assay
In the improved PCR-RFLP assay, a 322-bp DNA band was amplified in theory in all genotypes (Fig. 2). Because the amplification product in the unaffected dog had 1 recognition site of MseI, it was cleaved into 2 fragments (i.e., 194- and 126-bp bands) after digestion. In the affected dog, because an additional restriction site due to the mutation was designed to be in the midst of the 126-bp fragment, the amplified band was cleaved into apparently 2 fragments (i.e., 194- and two 62-bp bands) after the digestion. The amplification product in the carrier was cleaved into apparently 3 fragments of 194-, 126-, and two 62-bp bands. Thus, the result of the improved PCR-RFLP assay provided simpler and clearer genotype discrimination compared to that in the conventional PCR-RFLP assay.
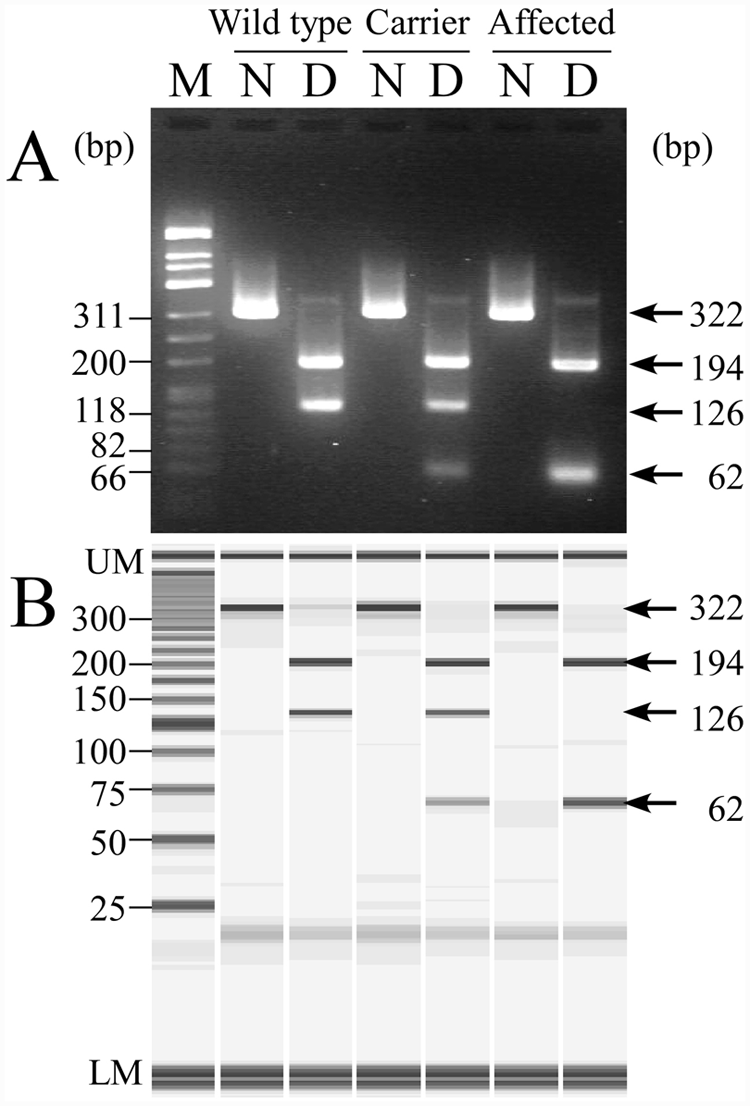
Neuronal ceroid lipofuscinosis genotyping of wild-type, heterozygous carrier and affected dogs by the improved polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) assay using agarose gel electrophoresis (
PCR-PIRA assay
In the PCR-PIRA assay, a 29-bp sequence corresponding to the forward primer was cut from the amplified 290-bp band after the digestion of AflII, producing a 257-bp digested band in the mutant allele (Fig. 3). According to this theory, this assay produced a 290-bp undigested band in the unaffected dog, and 2 fragments (i.e., 290- and 257-bp bands) in the carrier. The 29-bp band was seen only on microchip electrophoresis in the affected dog because of its small band size and low density (Fig. 3A).
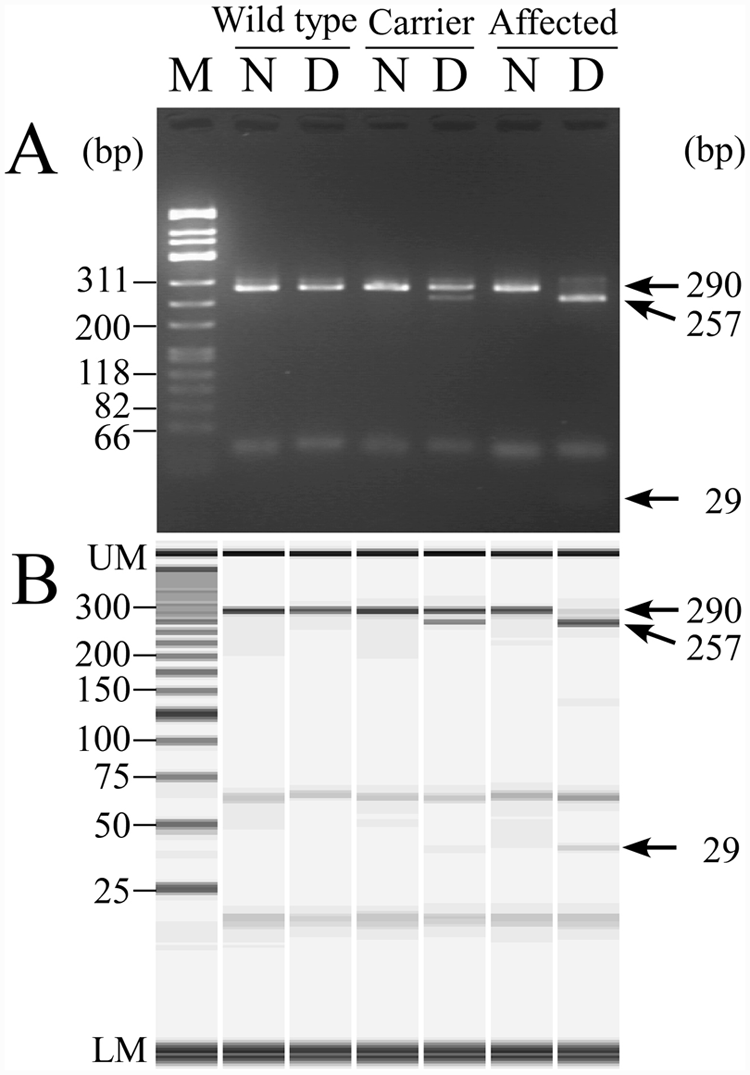
Neuronal ceroid lipofuscinosis genotyping of wild-type, heterozygous carrier and affected dogs by the polymerase chain reaction primer–induced restriction (PCR-PIRA) assay using agarose gel electrophoresis (
MS-PCR assay
As expected, in the MS-PCR assay, a 173-bp fragment was amplified with a wild allele-specific forward primer (49 mer) and a common reverse primer in the unaffected dog, and a 144-bp fragment with the mutant allele-specific forward primer (20 mer) in the affected dog (Fig. 4). In the carrier, these 2 bands of similar intensity were clearly seen on both agarose gel and microchip electrophoreses. In the gel image of microchip electrophoresis, a nonspecific band appeared in the midst of normal and mutant bands in the carrier (Fig. 4B), but did not interfere with discrimination of the carrier genotype.
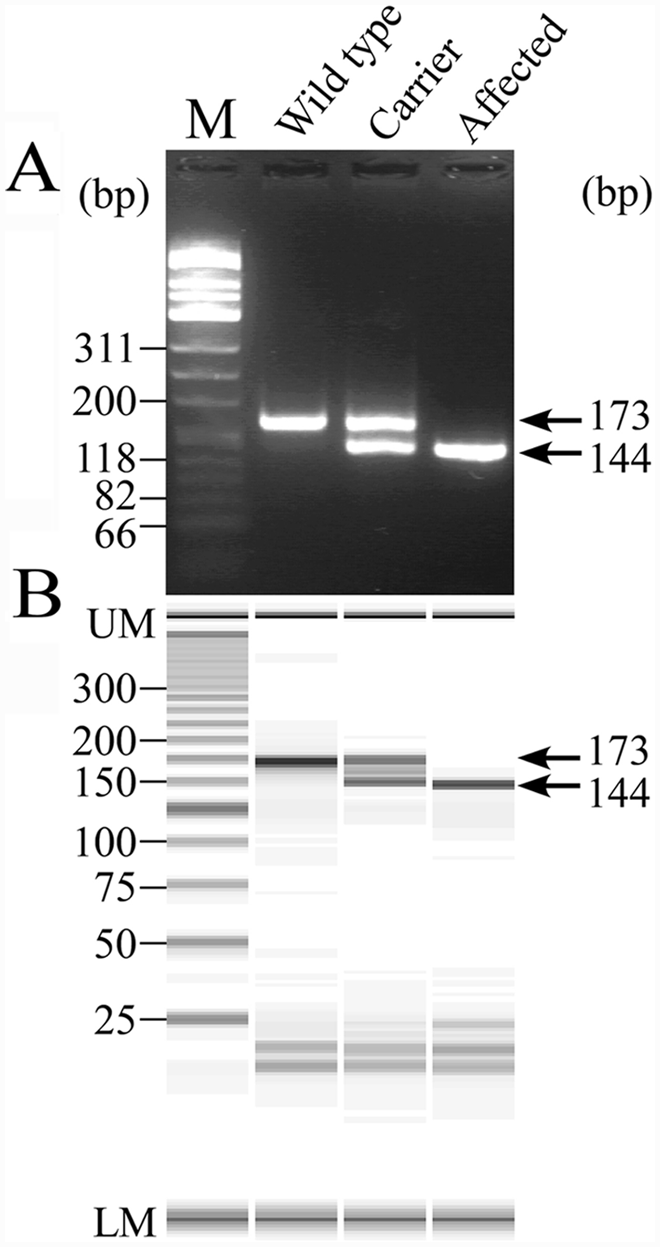
Neuronal ceroid lipofuscinosis genotyping of wild-type, heterozygous carrier and affected dogs by the mutagenically separated polymerase chain reaction (MS-PCR) assay using agarose gel electrophoresis (
Real-time PCR assay
Real-time PCR assay with TaqMan MGB probes clearly determined all the genotypes of NCL in Border Collies without any nonspecific allelic amplification (Fig. 5). Forty cycles of amplification were sufficient for genotyping using either blood or saliva DNA samples on FTA cards. The total required time for 40-cycle amplification was within 40 min. In addition, an allelic discrimination plot was constructed based on the 3 genotypes of amplification plots obtained using DNA samples from 46 Border Collies including 19 noncarrier, 19 carrier, and 8 affected dogs (Fig. 6). Three genotypes of the 46 dogs were clearly determined by this allelic discrimination plot and the results were completely consistent with those of other PCR-based assays developed in the present study.
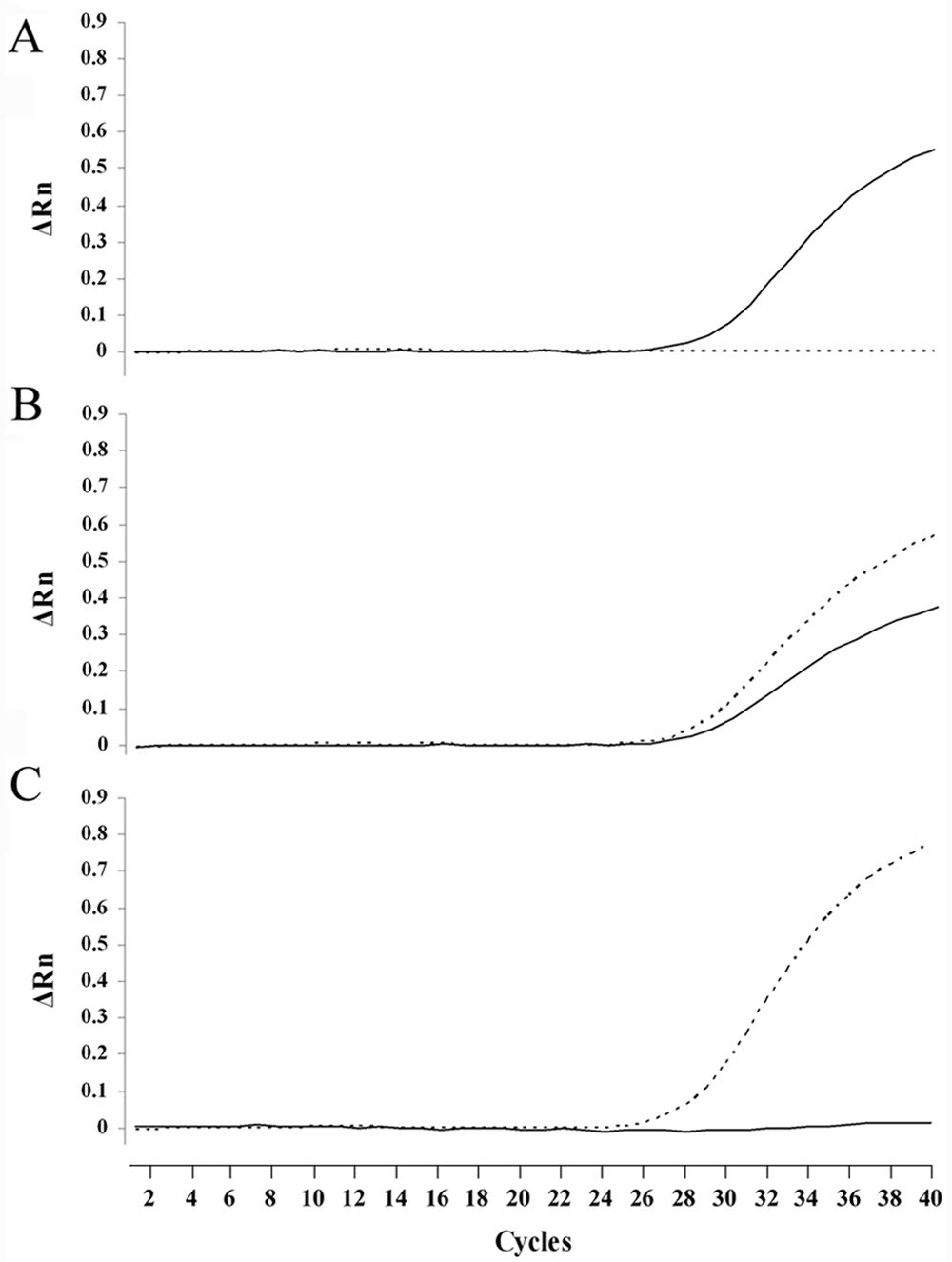
Real-time polymerase chain reaction amplification plots of wild-type and mutant alleles in canine neuronal ceroid lipofuscinosis. Amplification was plotted as fluorescence intensity (ΔRn value) against cycle number. The ΔRn value is the reporter dye signal normalized to internal reference dye and corrected for the baseline signal established in the first few cycles of reaction. Each of 3 amplification plots showed the normal (
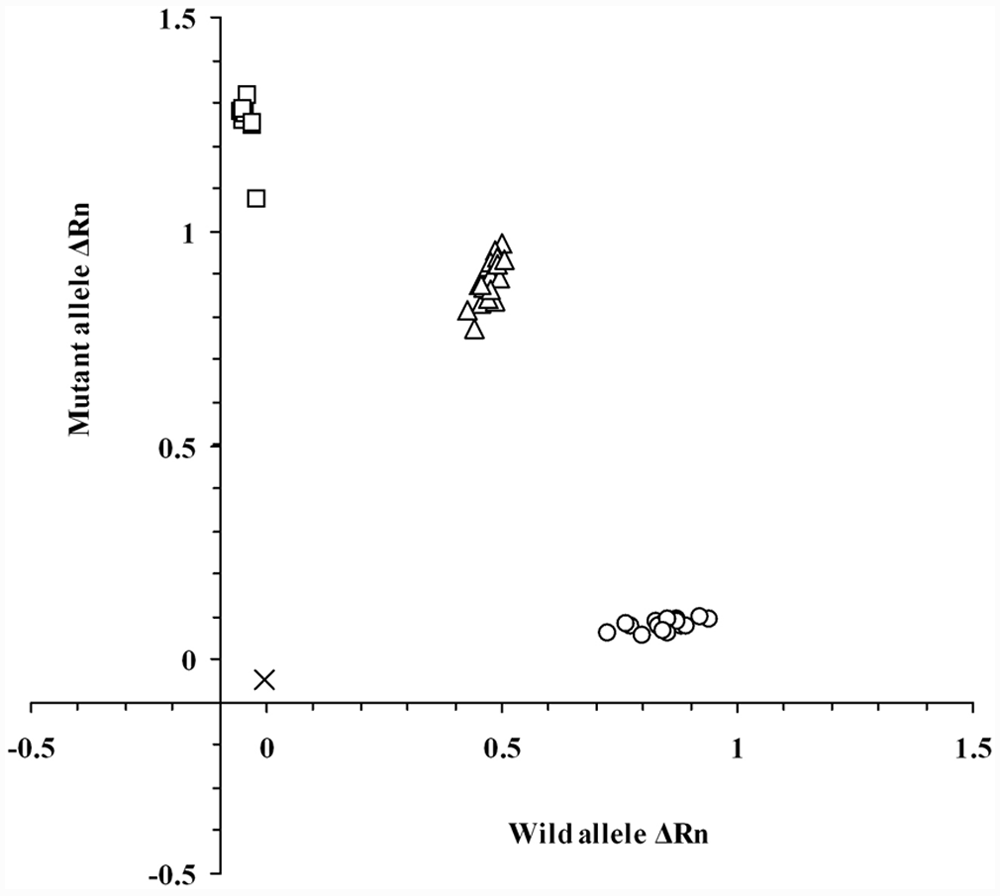
Allelic discrimination plot of end-point fluorescence real-time polymerase chain reaction data showing the 3 genotypes of canine ceroid lipofuscinosis. Allelic discrimination plot was depicted using representative 46 DNA samples in Border Collie dogs that had already been genotyped by other assays developed in the present study. The plot is expressed as fluorescence intensities (ΔRn value) for each allele at the X- and Y-axes. The ΔRn value in this figure is the end-point reporter dye signal normalized to internal reference dye and corrected for the baseline signal established in the first few cycles of reaction. x = no template control; ○ = normal genotype (19 samples); Δ = carrier genotype (19 samples); □ = affected genotype (8 samples).
Allele frequency
In the genotyping survey carried out on 407 Border Collies in Japan, 33 dogs were heterozygous carriers, indicating that the carrier frequency was 8.1%. However, there were no affected dogs detected in this random survey, resulting in an allele frequency of 0.041.
Discussion
In recessive inherited diseases such as lysosomal diseases, carriers that have one abnormal allele in the gene pair but have a normal clinical appearance are the most important genotype to identify because, although there are no physical clues to the presence of disease in these animals, the pathogenic mutation is transmitted to half of their progeny. 4 The frequency of carriers in a population substantially exceeds the incidence of affected individuals and thus the carrier state makes recessive disease the most dangerous of all patterns of inheritance in purebred animals. 31 Therefore, to prevent and eradicate fatal hereditary diseases, the determination of genotypes through systematic monitoring surveys and continuous removal of carriers from breeding colonies might be the most important and efficient measures. In the present study, rapid and reliable assays were developed to determine the genotypes of NCL in Border Collies and the current allele frequency in Japan was surveyed to examine the necessity of disease control.
The conventional genetic assay reported previously 17 was not a reliable method for easily discriminating the genotypes because of the complex and unclear DNA fragmentation (Fig. 1). In the improved PCR-RFLP assay, the mutant homozygote could be easily discriminated from the noncarrier and carrier genotypes because a 126-bp fragment was absent in affected dogs only, but, this assay showed a lower reliability in discriminating between noncarrier and carrier genotypes because the judgment depends on the absence or presence of a slightly weak 62-bp band (Fig. 2). In the PCR-PIRA assay, it was relatively easy to discriminate between carrier and other genotypes because the carrier genotype clearly had double DNA fragments of 290- and 257-bp (Fig. 3). However, the difference between these 2 band sizes was only 33-bp, making it slightly difficult to discriminate between normal and affected genotypes especially on agarose gel electrophoresis (Fig. 3A), but the judgment was easier using microchip electrophoresis (Fig. 3B). The MS-PCR assay clearly demonstrated the genotypes in a single tube without any restriction endonuclease digestion unlike the PCR-RFLP and PCR-PIRA assays (Fig. 4). Therefore, the MS-PCR assay was more rapid, simpler, and less expensive than PCR-RFLP and PCR-PIRA assays.
Microchip electrophoresis has recently attracted considerable attention in DNA analysis because of its high efficiency, high throughput, timesaving ability, easy operation, and low consumption of samples and reagents.6,21,33 The use of microchip electrophoresis can markedly shorten the time for analysis of DNA fragment patterns, which is approximately 3 min per sample. In the present study, the discrimination of genotypes was more rapid and clearer using microchip electrophoresis rather than agarose gel electrophoresis because of its high resolution and detection sensitivity (Figs. 2B, 3B, and 4B). On MS-PCR assay, a nonspecific band appeared in the midst of normal and mutant bands in the carrier (Fig. 4B). This was regarded as heteroduplex of PCR products for wild-type and mutant alleles. This band might be detected by the high sensitivity of the microchip electrophoresis, but it is less of an obstacle to the judgment of carrier genotype.
Recently, rapid real-time quantitative PCR approaches have been developed to detect mutations in genes causing hereditary diseases in human beings13,30 and animals.5,6 Real-time PCR is generally considered more sensitive, rapid, and less time-consuming than PCR in combination with electrophoresis analysis.5,6,13,30 In particular, real-time PCR assay in combination with FTA cards for sampling can markedly shorten the time required for genotyping and simplify the procedure. 5 In the present study, therefore, the application of real-time PCR with TaqMan MGB probes was investigated to establish a rapid and reliable genotyping technique for NCL in Border Collies. As a result, this assay clearly determined all genotypes (Fig. 5). In addition, an allelic discrimination plot analysis yielded multiple results simultaneously (Fig. 6), which can be particularly useful for large-scale epidemiological survey and preventive screening as well as diagnosis of affected dogs.
In domestic animals, there are few reports of the carrier frequency in fatal inherited diseases. In a preliminary genotyping survey carried out in Shiba dogs on a single mutation of GM1 gangliosidosis, one of the lysosomal storage diseases, the carrier frequency in Japan was reported to be 2.9%. 32 In mixed breed cats in Japan, it is reported that the allele frequency for a single mutation of GM2 gangliosidosis variant 0 is less than 0.1%. 21 In the present study, the carrier frequency of the canine CLN5 single mutation (c.619C>T) was 8.1% in the population of Border Collies in Japan. Although there is no objective criterion to evaluate the degree of frequency, the carrier frequency of NCL in Border Collies in Japan may be very high compared with those in other human and animal lysosomal diseases. This high carrier frequency suggests the presence of a founder effect in the population of Border Collies in Japan.
In conclusion, the present study demonstrates that 4 types of genotyping assays including improved PCR-RFLP, PCR-PIRA, MS-PCR, and real-time PCR are useful for diagnosis and screening of NCL in Border Collies. In particular, real-time PCR assay with TaqMan MGB probes is suitable for large-scale preventive screening. Using these assays, measures to prevent the continuing spread of this fatal inherited disease should be undertaken in the population of Border Collies.
Footnotes
Acknowledgements
The authors are grateful to all members of the Japan Border Collie Health Network for their help with the collection of DNA samples of Border Collies and to all dog owners who gave approval for the scientific evaluation of their dogs’ DNA.
a.
FTA classic card, Indicating FTA Classic Card, Harris Uni-Core Punch (size 1.2 mm), FTA Purification Reagent; Whatman International Ltd, Piscataway, NJ.
b.
10×TE Powder (pH 8.0), Wako Pure Chemical Industries Ltd., Osaka, Japan.
c.
GoTaq Hot Start Green Master Mix, Promega Corp., Madison, WI.
d.
MseI, 10× NE Buffer, 100x BSA, AflII; New England Biolabs, Ipswich, MA.
e.
Certified Low Range Ultra Agarose; Bio-Rad Laboratories, Hercules, CA.
f.
øX174/Hinf I digest, Nippon Gene, Tokyo, Japan.
g.
MCE-202 MultiNA Microchip Electrophoresis System, DNA-500 Kit; Shimadzu Corp., Kyoto, Japan.
h.
SYBR Gold Nucleic Acid Gel Stain, 25 bp DNA Ladder; Invitrogen Corp., Carlsbad, CA.
i.
DNA Extract All Lysis Reagents Kit, StepOne Real-Time PCR System, TaqMan GTXpress Master Mix, TaqMan SNP Genotyping Assays, StepOne software (version 2.0); Applied Biosystems, Foster City, CA.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This study was supported financially by grants (nos. 20380173, 20-08112, and 21658109, OY) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.