
Abstract
Reverse transcription loop-mediated isothermal amplification (RT-LAMP) was developed for detecting Infectious pancreatic necrosis virus (IPNV) in chum salmon (Oncorhynchus keta) in Korea. The RT-LAMP is a novel approach of nucleic acid gene amplification with high specificity, sensitivity, and rapidity under isothermal conditions. Based on the VP2/NS gene sequence of VR-299 and Jasper strains, a set of 6 IPNV-specific primers was designed to recognize 8 diverse sequences of the IPNV RNA. The assay was successfully optimized to detect IPNV at 65°C in 30 min. The detection limit was 0.075 tissue culture infectious dose infecting 50% of inoculated cultures per milliliter (TCID50/ml) from IPNV-infected rainbow trout gonad (RTG)-2 cells, whereas nested reverse transcription polymerase chain reaction (nRT-PCR) had a sensitivity of 7.5 TCID50/ml. Using RT-LAMP assay, field samples were analyzed and the results compared with those of nRT-PCR assay. Two hundred and sixty-six out of 659 (40.4%) samples were IPNV-positive by RT-LAMP, whereas 182 of 659 samples (27.6%) were IPNV-positive by nRT-PCR. The results indicate that RT-LAMP can be a useful tool for early field diagnosis of IPNV.
Introduction
Infectious pancreatic necrosis (IPN) is a highly infectious viral disease of salmonids considered as one of the most serious limiting factors for salmonid aquaculture. 18 The disease has a wide geographical distribution, occurring in most of the salmonid farming countries of North and South America, Europe, and Asia. 3,11,18 Clinical signs include darkening of the body, mild swelling on the head, abdominal distension, and a spiral swimming motion. 18 Infectious pancreatic necrosis virus (IPNV) can cause higher than 90% mortality in rainbow trout (Oncorhynchus mykiss) fry depending on several factors such as virus strain, host age, and stocking density. Significant mortalities due to IPNV infection also occur in the post-smolt stage of Atlantic salmon (Salmo salar) after sea transfer. 1,8,18,20 Asymptomatic survivors can act as carriers of viable IPNV for life and can shed infective viral particles, which can be transmitted to their offsprings. 20
Classification of IPNV strains has been traditionally performed by serological techniques. Most IPNV strains belong to serogroup A, which includes 9 serotypes, serotypes A1 (reference strain, West Buxton [WB]), A2 (Spajarup [Sp]), A3 (Abild [Ab]), A4 (Hecht [He]), A5 (Tellina [Te]), A6 (Canada 1 [C1]), A7 (Canada 2 [C2]), A8 (Canada 3 [C3]), and A9 (Jasper [Ja]), 18 whereas only a few IPNV strains comprise serogroup B (B1, type strain TV-1). 6 Recent phylogenetic analysis of IPNV based on the sequencing analysis of the VP2 gene 2,10 revealed that 6 genogroups (I–VI) including 10 genotypes (I.1, I.2, II, III.1, III.2, IV.1, IV.2, V.1, V.2, VI) exist. 2 Most IPNV isolates from Europe are identified as Sp (genogroup V) and Ab (genogroup II) strains, whereas most of the isolates from salmonids in Asia and North America are classified as Ja (genotype I.1) and VR-299 (genotype I.2) strains. 2,7,10,11 In Korea, IPNV has been isolated from chum salmon (Oncorhynchus keta), rainbow trout, goldfish (Carassius auratus), and Korean rockfish (Sebastes schlegelii), all of which are known to belong to VR-299. 5,9,11
The reverse transcription polymerase chain reaction (RT-PCR) assay has been widely used for detection and identification of salmonid birnaviruses. 13,23,24 However, it requires a second amplification step that is more prone to contamination, and it is also time consuming. A novel method, loop-mediated isothermal amplification (LAMP) assay, was recently introduced for improving the diagnosis of several aquatic animal diseases. 19 First developed in the year 2000, the LAMP can amplify nucleotides from a few copies to 109 copies under isothermal conditions within 1 hr. 16 The principle of LAMP is based on autocycling strand displacement DNA synthesis by the Bst DNA polymerase large fragment, which has high strand displacement activity, with 2 specially designed inner and 2 outer primers. 16 The LAMP is highly specific because the target sequence can be detected by 6 independent sequences in the initial stage followed by 4 independent sequences in the later stages of the LAMP reaction. As such assay is held under isothermal condition, it can be easily completed in a water bath or a heating block; thus, an expensive thermal cycler is not necessary.
The LAMP assay is also applicable for detecting RNA by using a reverse transcription (RT) together with DNA polymerase. 16 The RT-LAMP assay was developed to detect many fish pathogens, 19 which include Infectious hematopoietic necrosis virus (IHNV), 4,14 Infectious salmon anemia virus (ISAV), 14 Viral hemorrhagic septicemia virus (VHSV), 21 and Spring viraemia of carp virus (SVCV). 12 The sensitivity of RT-LAMP assay was greater than that of the RT-PCR assay in these studies. The RT-LAMP has been previously used for detecting IPNV, 22 and the primers used were designed on the basis of the NS/VP3 encoding region of the Sp and Ab strains, which are the most common strains in Europe. 22 In the current study, the RT-LAMP was developed for detecting IPNV based on sequences of major strains in Asia-Pacific regions (VR-299 and Ja strains). Moreover, the detection limits of IPNV between the RT-LAMP and nRT-PCR assays were compared, and its applicability to field samples was assessed.
Materials and methods
Viruses
Infectious pancreatic necrosis virus (Ja, VR-299, Sp, and Ab strains) and VHSV were propagated in rainbow trout gonad (RTG)-2 cells. Infectious hematopoietic necrosis virus was propagated in Chinook salmon embryo (CHSE)-214 cells. Virus-infected cells were grown in minimum essential medium (MEM) a supplemented with 10% fetal bovine serum (FBS), b 200 mM L-glutamine, 50 IU/ml of penicillin, and 50 mg/ml of streptomycin, and maintained at 15°C. Then, the viruses were titrated in 96-well plates, and the tissue culture infectious dose infecting 50% of inoculated cultures per milliliter (TCID50/ml) was calculated. 17
RNA extraction
Wild adult chum salmon (1–4 kg body weight) and fry (0.5–0.9 g body weight) with no clinical signs of disease were collected and used for testing. One hundred milligrams of fresh kidney and spleen were homogenized in 750 µl of total RNA isolation reagent c and incubated at room temperature for 5 min. Then, 200 µl of chloroform was added to the homogenates and mixed by vortex. The suspension was incubated at room temperature for 10 min and centrifuged at 12,000 × g for 10 min. The upper solution was transferred into a new 1.5-ml microcentrifuge tube and precipitated by adding 200 µl of 100% isopropanol on ice for 10 min, then centrifuged at 12,000 × g for 10 min. The RNA pellet was washed with 70% (v/v) ethanol, centrifuged at 9,000 × g for 5 min, and then dried at 95°C in the incubator for 2 min or until the ethanol was evaporated. To elute RNA, diethylpyrocarbonate (DEPC)-treated water d was added to make a final concentration of 100 ng/µl and kept at –80°C. The RNA quantification was measured by spectrophotometer. e
Genomic viral RNA was extracted from 500 µl of IPNV-infected cell culture supernatant as described above and used for optimizing the RT-LAMP conditions. After elution of the RNA in DEPC-treated water, it was stored at −80°C until required.
Primers for the RT-LAMP assay
Reverse transcription loop-mediated isothermal amplification primers for IPNV were designed according to consensus sequences of the IPNV VP2/NS gene (VR-299 and Ja strains; GenBank accession nos. AF342729 and M18049) using LAMP primer designing software. f The VP2/NS junction region was selected because it is known as a conserved region of aquatic birnaviruses, and because the VR299 and Ja strains are the most common strain of IPNV in Asia-Pacific regions. The forward inner primer (FIP) consisted of the complementary sequence of F1c (22 nucleotide [nt]), a TTTT spacer, and the sense sequence of F2 (18 nt). The backward inner primer (BIP) contained a sense sequence of B1c (20 nt), a TTTT spacer, and the complementary sequence of B2 (18 nt). The outer primers consisted of the F3 (20 nt) and complementary sequence of B3 (18 nt). Loop primers were designed in order to increase the LAMP sensitivity and contained a loop forward primer (LF; 19 nt) and loop backward primer (LB; 19 nt). The details of the primers are listed in Table 1.
Oligonucleotide primers developed for detecting Infectious pancreatic necrosis virus by reverse transcription loop-mediated isothermal amplification assay.*

F3 = outer primer consisting of 20 nucleotide (nt); B3 = outer primer of complementary sequence consisting of 18 nt; FIP = forward inner primer; BIP = backward inner primer; LF = loop forward primer of 19 nt; LB = loop backward primer of 19 nt.
Conditions of the RT-LAMP reaction
The RT-LAMP was carried out in 25 µl of total reaction mixture containing 2 µM each FIP and BIP, 0.2 µM each F3 and B3, 2 µM each LF and LB primers, 1× thermopol-supplied reaction buffer, 0.8 M betaine, a 8 mM magnesium sulfate, a 1 mM each deoxynucleotide triphosphate mix, g 6 U of Bst DNA polymerase, h and 0.25 U of Avian myeloblastosis virus reverse transcriptase. i Uninfected RTG-2 cells were included as negative control. The reaction temperature and time were optimized at 65°C for 30 min to make specific and rapid amplification of IPNV. The RT-LAMP products were analyzed by 2% gel electrophoresis.
Nested RT-PCR assay for Infectious pancreatic necrosis virus detection
The RT-PCR assay was conducted by adding P-1 and P-2 primers 23 (Table 2) and 100 ng/µl of total RNA into the lyophilized RT-PCR premix tube, d to a reaction volume of 20 µl with DEPC-treated water. Reverse transcription amplification was conducted under the initial step at 42°C for 45 min, then 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 48°C for 30 sec, and 72°C for 30 sec. A final extension step was conducted at 72°C for 5 min. The expected PCR product was 359 base pairs (bp). A nested PCR amplification was carried out by mixing 5 µl of RT-PCR products with 10 µM of each P-3 and P-4 primers 23 (Table 2) in the PCR premix tube, d then water was added up to a final volume of 20 µl. The expected size of PCR product was 168 bp. The following amplification conditions were used an initial denaturation step at 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 48°C for 30 sec, 72°C for 35 sec, and a final extension step was conducted at 72°C for 5 min.
Oligonucleotide primers used for detecting Infectious pancreatic necrosis virus by nested reverse transcription polymerase chain reaction assay.
Sensitivity and specificity of RT-LAMP and sensitivity of the nested RT-PCR
To determine the sensitivity of the RT-LAMP and nRT-PCR assays, 10-fold serial dilutions (7.5 × 104 TCID50/ml to 7.5 × 10−3 TCID50/ml) of RNA extracted from IPNV (Ja strain)-infected RTG-2 cells were tested with the optimized conditions for the RT-LAMP and the nRT-PCR as described above. The specificity of the RT-LAMP assay was tested using total RNA extracted from IPNV strains (VR299, Ab, and Sp), VHSV-infected RTG-2 cells, and IHNV-infected CHSE-214 cells, respectively.
Evaluation of the RT-LAMP assay with field samples
Comparing the field sample detection results of the RT-LAMP with those of the nRT-PCR allowed researchers to assess the feasibility of the RT-LAMP assay to detect IPNV. The fry and adult chum salmon were collected from the Namdae River basin and hatcheries at Yangyang City on the east coast of Korea during 2006–2010 for routine monitoring of IPNV. The kidney and spleen from individual adult fish and pooled 5 whole fry were used for RNA extraction as mentioned above. Then, RT-LAMP and nRT-PCR assays were conducted with the conditions above and the results were compared.
Sequencing and sequence alignment
The LAMP products were purified by gel purification kit d according to the manufacture’s protocol. Products were then digested using MspA1I enzyme h at 37°C for 16 hr. The MspA1I enzyme-digested fragment of 177 bp was produced as predicted from the amplicon structure. After repurification, 100 ng of the digested products were cloned into a vector. i Plasmid DNA was subsequently purified using a commercial kit j according to the manufacturer’s protocol. A 50 ng/µl of purified plasmid DNA was then directly sequenced k by using M13 universal primer set. The sequences of IPNV VP2/NS junction region were aligned with the sequences available in the GenBank database (National Center for Biotechnology Information) according to the ClustalW software. 25
Results
Sensitivity of RT-LAMP and nested RT-PCR and specificity of RT-LAMP detection
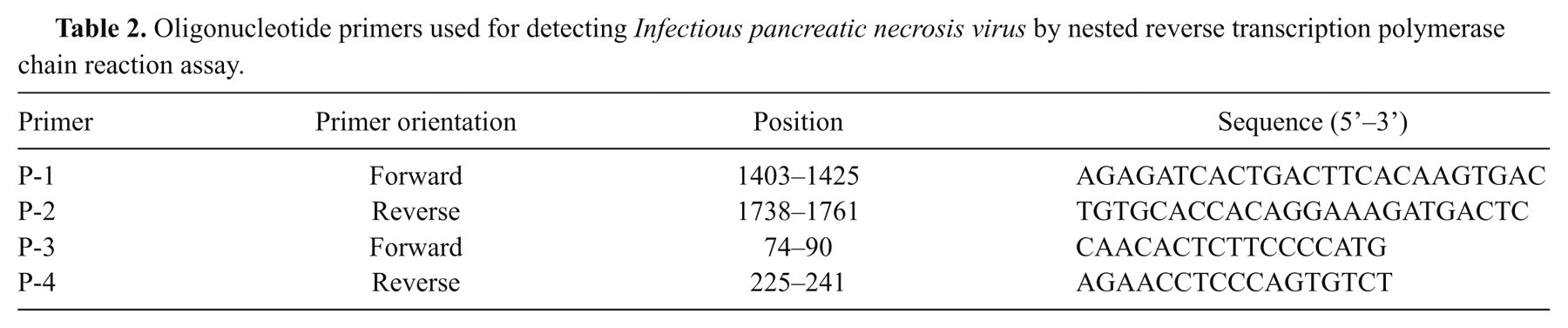
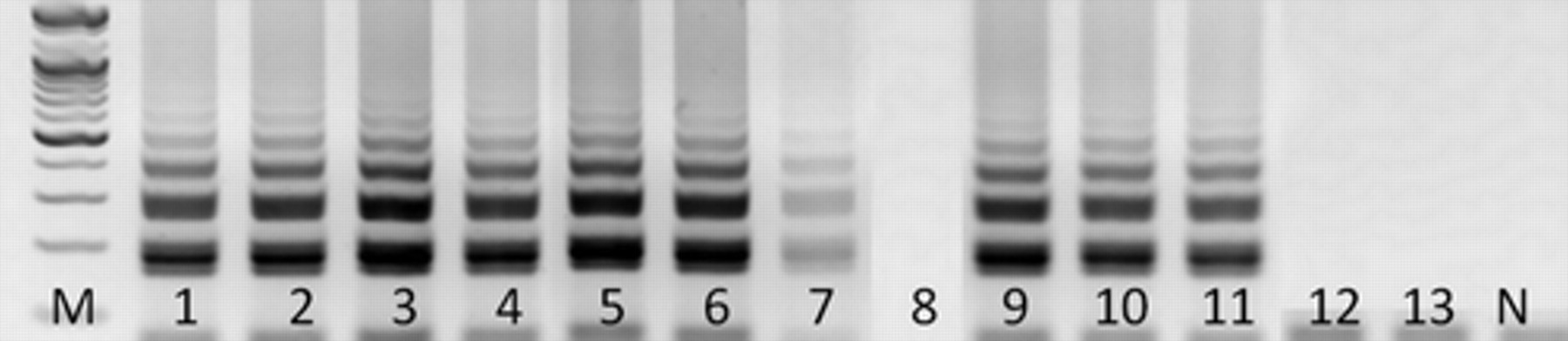
The sensitivity of RT-LAMP was 100-times higher than the nRT-PCR. The RT-LAMP was able to detect the template at 0.075 TCID50/ml (Fig. 1, lane 7), whereas the nRT-PCR detected the template at 7.5 TCID50/ml (Fig. 2, lane 5). The cross amplification of RNA of each salmonid virus (i.e., IPNV-Ja, 7.5 × 108 TCID50/ml; VR299, 7.6 × 108 TCID50/ml; Ab, 7.4 × 108 TCID50/ml; Sp, 7.2 × 108 TCID50/ml) strains–infected RTG-2 cells, VHSV-infected RTG-2 cells (7.7 × 108 TCID50/ml), and IHNV-infected CHSE-214 cells (7.5 × 108 TCID50/ml) with the RT-LAMP primers was also used for confirming the specificity of the RT-LAMP assay. All IPNV isolates were positive, but none of the other salmonid fish viruses was positive with the primers, indicating that the RT-LAMP assay is specific to IPNV (Fig. 1, lanes 9–13).
The sensitivity and specificity of reverse transcription loop-mediated isothermal amplification assay for detection of Infectious pancreatic necrosis virus (IPNV). Lane M: 100-bp DNA marker; lanes 1–8: 10-fold dilutions of RNA extracted from IPNV (Jasper strain)-infected rainbow trout gonad (RTG)-2 cells (7.5 × 104 TCID50/ml to 7.5 × 10−3 TCID50/ml); lane 9: IPNV (Abild strain)-infected RTG-2 cells; lane 10: IPNV (VR-299 strain)-infected RTG-2 cells; lane 11: IPNV (Spajarup strain)-infected RTG-2 cells; lane 12: Viral hemorrhagic septicemia virus (VHSV)-infected RTG-2 cells; lane 13: IHNV-infected Chinook salmon embryo (CHSE-214) cells; N = negative control.
The sensitivity of nested reverse transcription polymerase chain reaction for detection of Infectious pancreatic necrosis virus (IPNV). Lane M: 100-bp DNA marker; lanes 1–8: 10-fold dilutions of RNA extracted from IPNV (Jasper strain)-infected rainbow trout gonad (RTG-2) cells (7.5 × 104 TCID50/ml to 7.5 × 10−3 TCID50/ml); N = negative control.
Evaluation of the RT-LAMP method with field samples
The prevalence of IPNV in chum salmon is summarized in Table 3. The occurrence of IPNV in chum salmon was 266 out of 659 (40.4%) by the RT-LAMP. In contrast, the nRT-PCR results revealed the prevalence was 182 out of 659 (27.6%). All nRT-PCR–positive samples were RT-LAMP positive in each group and each year.
Comparison of the prevalence of Infectious pancreatic necrosis virus in chum salmon (Oncorhynchus keta) by nested reverse transcription polymerase chain reaction (nRT-PCR) and reverse transcription loop-mediated isothermal amplification (RT-LAMP) assays.*
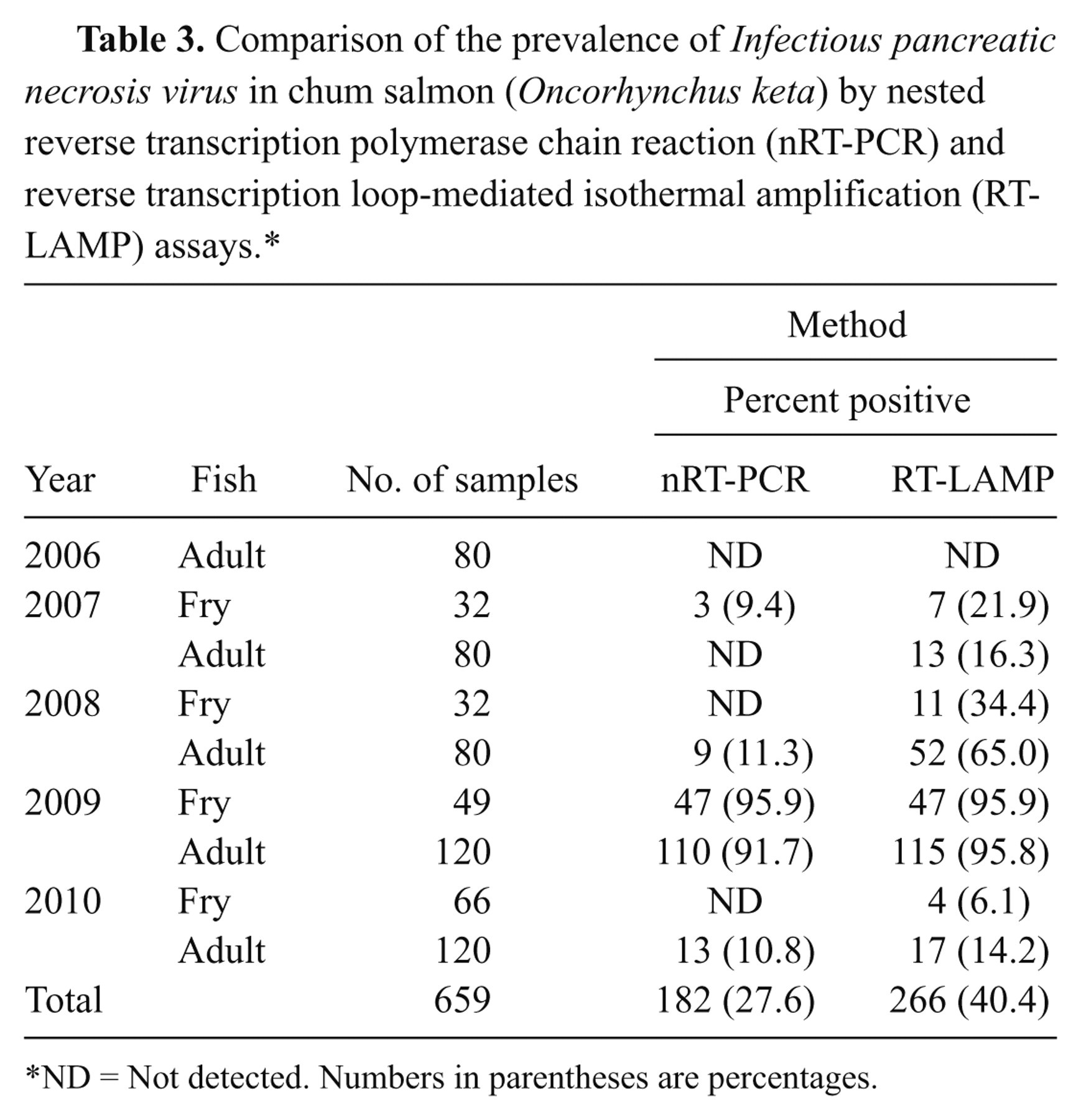
ND = Not detected. Numbers in parentheses are percentages.
Sequence alignment
Ten different RT-LAMP products of IPNV were sequenced and aligned with published sequences of the VR-299 (accession no. AB343572), Ja (accession no. M18049), Ab (accession no. AF342729), and Sp (accession no. L13988) strains available in the GenBank database. All the sequences had a high degree of homology with the VR-299 and Ja strains (Fig. 3). In particular, LAMP products of 5 samples (062, 072, 073, 082, and 091) were completely homologous with the VR-299 and Ja strains; only 4 nucleotides differences (out of 177) were observed in LAMP products of 5 samples (061, 071, 081, 083, and 092).

Sequence alignment of 10 different the Infectious pancreatic necrosis virus reverse transcription loop-mediated isothermal amplification assay products with the reference strains. Reference strains: VR299: AB343572, Jasper: M18049, Abild: AF342729, and Spajarup: L13988. A dot in each sequence represents a nucleotide identical to that of the VR299 strain.
Discussion
The loop-mediated isothermal amplification is a novel diagnostic tool to detect aquatic pathogens. 19 In the current study, the RT-LAMP reaction could be completed in 30 min, at 65°C, indicating the high amplification effectiveness of the LAMP assay.
The developed IPNV-specific RT-LAMP could detect not only Asian-Pacific strains, but also European strains (Sp and Ab strains) with the primers in the current study. In Korea, there is no report on the European strains at the present time. Therefore, the IPNV-specific primers developed in the current study can be useful for detecting the European IPNV strains (Sp and Ab) and helpful for preventing the potential introduction of those strains. But, it is still unclear if the RT-LAMP primers in the present study can detect other IPNV genogroups because they were not tested. In addition, the IPNV-specific RT-LAMP primers could not detect other salmonid viruses such as IHNV and VHSV. The increased sensitivity of the RT-LAMP compared to the nRT-PCR in the current study was because it used 6 primers recognizing 8 distinct regions on the target RNA of IPNV. 15,16
The RT-LAMP generated products were sequenced to confirm the amplification of a correct target sequence by this assay. Sequences alignment showed that the RT-LAMP products have the genetic similarity with the VR-299 (98–100%) and Ja strains (98–100%). Because the RT-LAMP product sequences were too short for the comparison, the VR-299 and Ja strains could not be distinguished in the current study.
The increased sensitivity of RT-LAMP assay for detecting IPNV with field samples over the nRT-PCR assay indicates that the RT-LAMP assay developed in the current study is suitable for early field diagnosis. Moreover, the RT-LAMP assay developed is rapid, sensitive, cost-effective, and specific for detecting IPNV. Such assay needs only a water bath or a heating black for isothermal amplification and the result can be achieved within 1 h including gel electrophoresis, while nRT-PCR needs 4 hr to be completed. Considering the importance of salmonids aquaculture and frequent domestic translocation of the eggs and the fry in Korea, the RT-LAMP assay can be a suitable test for routine early field diagnosis and quarantine programs to prevent spread of infection.
Footnotes
Acknowledgements
The authors thank K. B. Seong and C. H. Lee (Cold Water Fish Research Center, Yangyang, Korea) for their help in collecting fish samples.
a.
Sigma-Aldrich, St. Louis, MO.
b.
Lonza, Walkersville, MD.
c.
TRI reagent, Molecular Research Center, Cincinnati, OH.
d.
Bioneer, Daejeon, Korea.
e.
NanoDrop 1000, Thermo Scientific, Wilmington, DE.
f.
PrimerExplorer V4, Eiken Chemical, Tokyo, Japan.
g.
Solgent, Seoul, Korea.
h.
New England Biolabs, Ipswich, MA.
i.
pGEM-T, Promega Corp., Madison, WI.
j.
Qiagen GmBH, Hilden, Germany.
k.
3730xl Genetic Analyzer, Applied Biosystems, Foster City, CA.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The current work was supported by a grant from the National Research Foundation of Korea (2009-0087136).