
Abstract
A SYTO9 real-time polymerase chain reaction assay for detection of pathogenic Leptospira spp. based on amplification of DNA gyrase subunit B (gyrB) gene has been optimized and evaluated for sensitivity and specificity on kidney and urine samples of New Zealand farmed deer. The detection limit was 103 cells/ml (2–10 copies/reaction). Comparison of the assay on deer kidneys (n = 268) with culture as the gold standard revealed a sensitivity and specificity of 85% and 99.2%, respectively. For deer urine (n = 113), the assay was compared with known inoculated samples and revealed a sensitivity and specificity of 96.7% and 100%, respectively. The assay was applied for quantifying pathogenic leptospires shed naturally in deer urine and revealed a detectable concentration of 3.7 × 103 to 1.7 × 106 cells/ml. To assess the assay’s capability for identifying pathogenic Leptospira spp., 14 field isolates of L. borgpetersenii serovar Hardjo-bovis and L. interrogans serovar Pomona were amplified for polymerase chain reaction (PCR) product, purified, and sequenced. When compared with the National Center for Biotechnology Information database, sequence data matched with L. borgpetersenii serovar Hardjo-bovis in 13 samples and L. interrogans serovar Pomona in 1 sample, which was consistent with the microscopic agglutination test (MAT). Sequence analysis of purified PCR product amplified directly from kidney and urine samples also yielded serovar-comparable MAT results. Results suggest that the assay is rapid, sensitive, and specific for detection of pathogenic leptospires in deer clinical samples. The developed assay can also be used for estimating the concentration of leptospires and identifying Leptospira spp. in combination with DNA sequencing.
Introduction
Leptospirosis is an emerging zoonotic disease with worldwide distribution. It is caused by infection with pathogenic Leptospira, helical-shaped motile spirochetes that belong to the family Leptospiraceae, genus Leptospira. Human beings and animals usually become infected by direct contact with blood, urine, or kidney of infected animals or indirectly by contact with surface water, mud, and soil that is contaminated with pathogenic leptospires from shedding animals. 20 There have been over 200 serovars of pathogenic Leptospira within 13 species isolated and described. 5
In New Zealand, 6 serovars within 2 species of Leptospira borgpetersenii and Leptospira interrogans have been isolated in animals. 2 Livestock, including farmed deer, play an important role in the spread of leptospirosis to both animals and human beings because livestock excrete organisms into the environment via urine, and at slaughter put meat workers at risk via urine and kidney tissue. Leptospirosis is a well-recognized clinical disease and subclinical infection in New Zealand farmed deer. 3,35 Leptospira borgpetersenii serovar Hardjo-bovis and Leptospira interrogans serovar Pomona are the most commonly detected serovars. 2
Culture of Leptospira organisms from clinical samples is the definitive method that allows identification of infecting serovars. However, culture is rarely used because it is technically demanding, tedious, complex, time consuming, prone to contamination, and expensive. The microscopic agglutination test (MAT) is the most widely used standard serology test because of its high sensitivity and specificity and its ability to identify leptospiral infection to the serogroup or serovar level. 26,33 However, MAT does not reflect the carrier status of the host. Microscopic agglutination test also has drawbacks including the maintenance of live Leptospira cultures for antigens and complexity of methods and interpretation that limits its use in the laboratory. 25,28 Conventional methods such as dark-field microscopy and Warthin–Starry staining to detect leptospires in clinical samples are unreliable because of their low sensitivity. 26
With the introduction of polymerase chain reaction (PCR) assays, rapid detection of small numbers of leptospires in clinical samples has become practical due to specific amplification of leptospiral DNA. The PCR assay can also determine the shedding and carrier status of leptospires, detecting leptospiral DNA in cattle urine, 4,7 pig kidney, 13 and sheep and goats semen and vaginal fluids. 22 There have also been applications of PCR for rapid diagnosis by detection of leptospires in blood. 14,29
Real-time PCR has further improved the diagnosis of leptospirosis. A new double-stranded DNA intercalating dye from the SYTO family has been introduced as an alternative to conventional SYBR Green I. The SYTO9 dye has been shown to produce robust and consistent DNA melting curves that are not affected by DNA concentration and can be used with a broad range of dye concentrations without causing PCR inhibition. It is easier to use than SYBR Green I, particularly for adapting conventional assays to a real-time format and for DNA melting curve analysis. 24
Recently, the DNA gyrase subunit B gene (gyrB) has been proposed as an alternative target to 16S ribosomal RNA gene for species identification. The gyrB gene is a single-copy gene, present in all bacteria, which encodes the ATPase domain of the enzyme DNA gyrase essential for DNA replication. The amino acid sequences of gyrB gene allow the comparison of bacterial taxonomy. 8 Recently, gyrB gene has been used as a target for identification of Leptospira species and is claimed as an alternate identification gene. 19,30
The current paper reports the evaluation and validation of a quantitative real-time PCR using SYTO9 detection technology in combination with amplification and sequencing of a partial fragment of gyrB gene for detection and identification of pathogenic Leptospira species in urine and kidney samples from New Zealand farmed deer. The detection limit, sensitivity, and specificity in clinical contexts were evaluated, examining kidney tissue and urine and comparing results with the gold standard, which is culture. The range of concentration of leptospires shed naturally in deer urine, identification of pathogenic Leptospira species based on DNA sequence of gyrB gene PCR product from field isolates of deer kidney and urine, and phylogenetic analysis is also reported.
Materials and methods
The Massey University Animal Ethics Committee under protocol 06/149 has approved all procedures involving manipulations on animals in the current study.
Sample sources
Deer kidneys (n = 268, range: 8–25 per farm) paired with blood samples, as available (n = 209, range: 0–25 per farm), were collected from 19 randomly selected slaughter lines from 18 farms at Deer Slaughter Premises (DSPs) in Feilding, Manawatu and Makarewa, Southland of New Zealand during the period of November 2006 to November 2008. Deer were rising 1-year-old, or older, and of both sexes. There were no data on previous exposure to Leptospira. Additionally, urine (n = 111, range: 15–28 per farm) collected from female rising 1-year-old deer on 5 commercial deer farms in the Manawatu and Hawke’s Bay regions of New Zealand in November 2007 as part of vaccine efficacy study were used in the study. Sampled animals were phenotypically red deer (Cervus elaphus) but possibly containing some wapiti (Cervus elaphus canadensis) genes.
Sample collection and preparation
Blood
Blood samples collected at DSPs were by free flow into a new 10-ml plain blood tube a after animal sticking, immediately after stunning, at the beginning of the slaughter line. On farms, blood samples were collected from the jugular vein of physically restrained deer using a new 20-gauge needle and a 10-ml plain blood tube. a Blood was held at 4°C before transport to the laboratory for centrifugation at 1,512 rcf for 15 min after which serum was aliquoted into new, labeled 1.5-ml microcentrifuge tubes and stored at –20°C.
Kidney
Whole kidneys were collected at the evisceration and inspection area of the slaughter board after capsule removal, put into labeled sterile plastic bags aseptically, and held at 4°C before transport to the laboratory. Within 6–24 hr of collection, the kidney surface was swabbed with 70% alcohol and randomly aspirated from several sites over the entire kidney into a 5-ml, sterile syringe using a sterile 16-gauge needle. Approximately 50 mg of the extracted kidney tissue was used for DNA extraction as determined by the kit. b The same amount was used for bacterial culture. Processed kidney samples were later split into two portions with half stored in freezer at –20°C and half stored in 10% neutral buffered formalin solution.
Urine
Urination was induced by administration of furo-semide c at 1−1.5 mg/kg intramuscularly. 12,34 A new 70-ml plastic collector was held beneath the vulva after urination began. As much middle-stream urine as possible was collected and immediately held at 4°C. After transport to the laboratory, urine was centrifuged at 1512 rcf for 10 min to provide sediment which was re-suspended with 400 µl of phosphate buffered saline (PBS) to neutralize the pH. 20 Half (200 µl) was used for DNA extraction as determined by the kit, b and half was used for bacterial culture. Leptospira-free deer urine was determined by no presence of organism or its DNA in bacterial culture and real-time PCR along with no detectable antibodies against Leptospira serovars Hardjo-bovis and Pomona in blood from the same animals.
Serology
The MAT was used to test serum reactivity to laboratory standardized Leptospira serovars Hardjo-bovis and Pomona. The method has been developed by the Leptospirosis Research Unit, Institute of Veterinary, Animal and Biomedical Sciences, Massey University (Palmerston, New Zealand) based on “Guidelines for the Control of Leptospirosis.” 11 A titer of ≥1:48 was considered positive for both serovars. 6,9
Bacterial culture
Five milliliters of Ellinghausen–McCullough–Johnson–Harris (EMJH) 10,17 was used as a selective medium with the addition of 100 µg/ml of 5-Fluorouracil d for contamination inhibition. 18 The processed samples (kidney and urine) were inoculated and subcultured into 2 consecutive tubes, incubated at 28−30°C, and examined every 2 weeks under dark-field microscopy for 4 months. Isolates of leptospires were serotyped against the standardized antisera of Leptospira serovars Hardjo-bovis and Pomona. The methods used were adapted from the standard protocols from the Massey University Leptospirosis Research Unit, based on “Guidelines for the Control of Leptospirosis.” 11
DNA extraction
The DNA was extracted from 50 mg of kidney tissue and from 200 µl of urine using a commercial kit b as per manufacturer’s instructions. The DNA was eluted in a final volume of 200 µl.
Polymerase chain reaction amplification
The real-time PCR assay was modified from a previously described assay. 30 Magnesium and primer titration was performed to determine the optimal concentration to give the lowest threshold cycle value. The SYTO9 e was used as a fluorescent, double-stranded DNA-specific, intercalating dye 24 for all real-time PCR assays. The assay was performed in a real-time genetic analyzer f using primers 2For 5’-TGAGCCAAGAAGAAACAAGCTACA-3’ and 504Rev 5’-MATGGTTCCRCTTTCCGAAGA-3’. 30 Each 25-µl reaction contained 2 µl of DNA extracted from samples, 1.5 µM SYTO9, 1× PCR buffer, 1.5 mM magnesium chloride, 200 µM of deoxyribonucleotide triphosphate (dNTP), 12.5 pmol of 2For primer, 12.5 pmol of 504Rev primer, 0.1% Fraction V bovine serum albumin, 1 U of Taq DNA polymerase, and double-distilled water. Thermal cycling consisted of initial denaturation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 10 sec, annealing at 60°C for 20 sec and extension at 72°C for 20 sec. Melting temperature (Tm) of PCR product was determined by melting curve analysis and performed by heating the PCR product from 70°C to 90°C and monitoring fluorescence change every 0.1°C. Confirmation of positive samples was determined by Tm of PCR product or performing electrophoresis of PCR product in a 1.5% agarose gel, stained with ethidium bromide, and visualized under ultraviolet light. The positive control used was Leptospira serovar Hardjo-bovis, and the negative control was double-distilled water.
Polymerase chain reaction product purification
The amplified PCR product was purified to remove excess primers and dNTP using a commercial kit g as per manufacturer’s instructions. Purified PCR product was eluted in a final volume of 50 µl.
DNA sequencing and phylogenetic analysis
The purified PCR products were forwarded to the Allan Wilson Centre Genome Service, Massey University, New Zealand for sequencing. Purified PCR products were sequenced in both orientations by the dideoxy-chain termination method using 2For- and 504Rev-specific primers. The sequence data were assembled and trimmed to a minimum of 2 contiguous sequences using the MT Navigator software. h Unknown sequence data were submitted to a nucleotide Basic Local Alignment Search Tool (BLAST; http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Analysis of DNA sequences was performed using the Geneious Basic version 4.6.1 software. i Multiple alignments of the DNA sequence were performed for phylogenetic comparison using the neighbor-joining method.
Detection limit
The limit of leptospiral DNA detection by real-time PCR was evaluated using artificially inoculated, Leptospira-free deer urine that was negative by culture and was from MAT-negative animals. Samples were prepared by mixing 180 µl of Leptospira-free deer urine with 20 µl of Leptospira serovar Hardjo-bovis (field isolate suspensions at a concentration of 2 × 108 cells/ml measured in a Petroff–Hausser counting chamber). 11 The next step was 7 serial ten-fold dilutions of 107–101cells/ml of Leptospira-free urine. The diluted samples were subsequently subjected to DNA extraction. Two microliters of extracted DNA samples obtained from all 7 dilutions were used as templates in the PCR. The procedure was repeated with 4 different field isolates of Leptospira serovar Hardjo-bovis for confirmation of detection limit.
Diagnostic sensitivity and specificity
Two experiments were carried out to determine the sensitivity and specificity of real-time PCR on kidney and urine samples. For kidneys, fresh tissue samples from 268 deer were subjected directly to real-time PCR and compared with culture as the gold standard using the techniques described above. Blood samples from 209 of those animals were subjected to MAT for serovar identification. The suitability of long-term storage of kidney tissue as either frozen or formalin-fixed samples for real-time PCR was compared with fresh samples from 11 kidneys. The agreement between results of the real-time PCR and kidney culture was tested by kappa statistic.
For urine, the sensitivity and specificity of real-time PCR was determined by creating artificially inoculated urine samples at the lowest detection limit (as described above) to mimic a natural gold standard. Thirty aliquots of 3 different field isolates of Leptospira serovar Hardjo-bovis containing approximately 2 × 108 cells/ml were diluted in PBS (pH 7.4) to 104 cells/ml. Thereafter, 20 µl of the diluted aliquot were inoculated with 180 µl of 30 different samples of Leptospira-free deer urine to achieve a concentration of 103 cells/ml. All samples were chilled at 4°C overnight to simulate the usual transport time for field-collected samples. All 30 inoculated urine samples and 83 Leptospira-free deer urine samples were subjected to DNA extraction and real-time PCR as described above to determine sensitivity and specificity.
Quantification of urinary shedding
Twenty-eight real-time PCR-positive deer urine samples from an on-farm vaccine efficacy study were used for estimating concentrations of leptospires shed naturally in deer urine. A standard curve was constructed for each PCR run from serial dilutions of leptospires inoculated in Leptospira-free deer urine, as described above. Each sample was tested in duplicate in each PCR run to estimate repeatability. The concentration of leptospires was calculated using the genetic analyzer software. f The concentration of leptospires was compared with culture from the same animals to determine the lowest concentration of leptospires in field-collected deer urine that could be cultured.
Identification of pathogenic Leptospira species
To assess ability to identify pathogenic Leptospira species, 14 isolates of unknown serovar from kidney and urine within the current study, 3 real-time PCR-positive deer kidney tissue samples, and 7 real-time PCR-positive deer urine samples were amplified for the product of gyrB gene. The amplified PCR products were then purified and forwarded to the Allan Wilson Genome Service Centre for DNA sequencing. The sequence data were BLAST searched to assess homologies with sequences in the National Center for Biotechnology Information (NCBI) database. Phylogenetic analysis was performed to compare the DNA sequence between isolates.
Results
Detection limit
The lowest detection limit was approximately 103 cells/ml or equivalent to 2–10 copies/reaction shown by both visualization of specific 500-bp DNA fragments in the gel (Fig. 1, lane 5) and melting curve analysis by real-time PCR. Such findings revealed that concentrations below 103 cells/ml could not be distinguished from the negative control (Fig. 2, line 5). The observed pattern was identical for all 5 different field isolates that were subjected to serial dilutions.
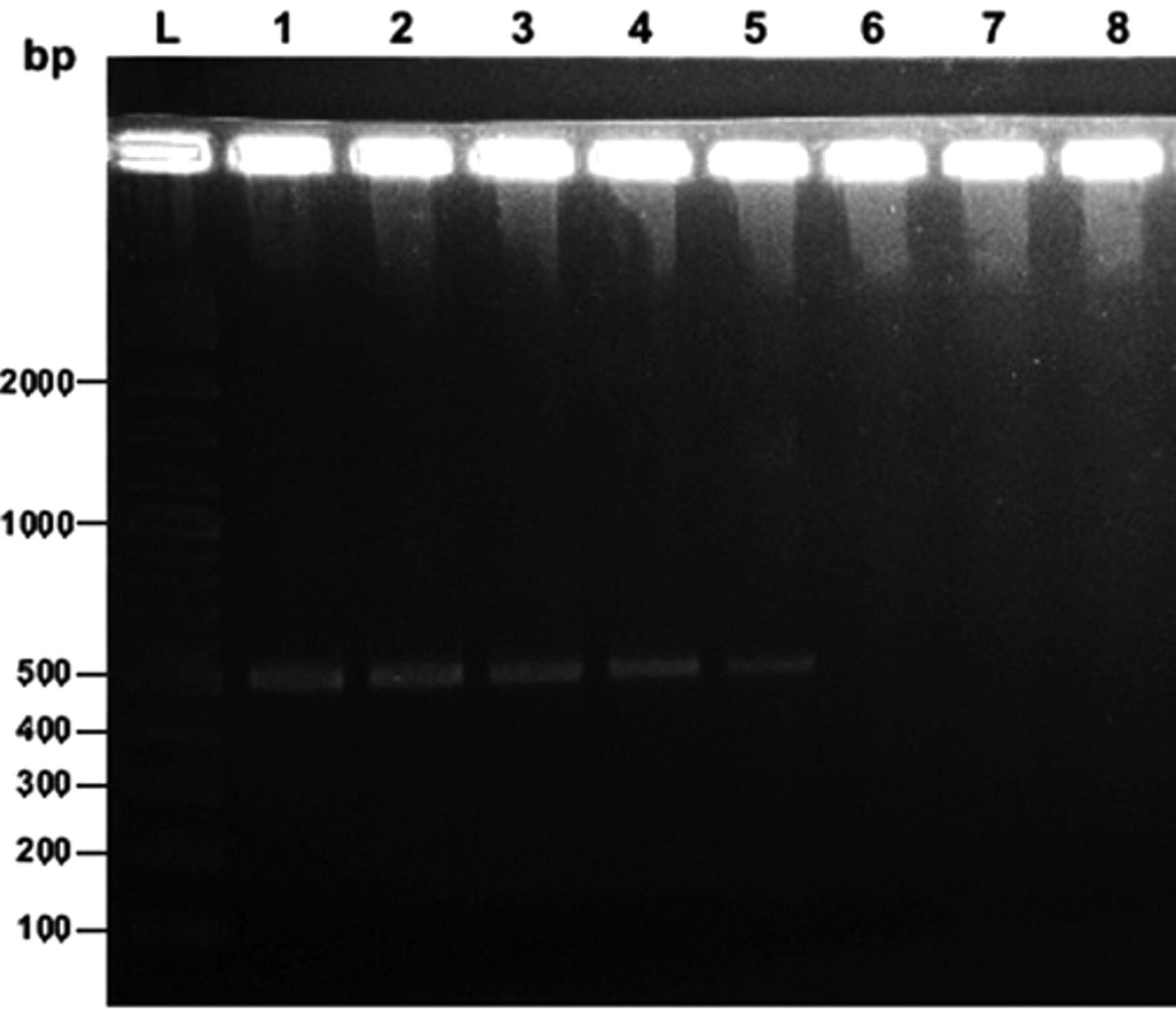
Representative gel electrophoresis showing the limit of detection (lane 5) of real-time polymerase chain reaction in Leptospira-free deer urine inoculated with various concentrations of a field isolate of Leptospira serovar Hardjo-bovis. Lane L: 2-log DNA ladder (range: 100–10,000 bp); lanes 1–7: 107–101 cells/ml, respectively; lane 8: negative control.
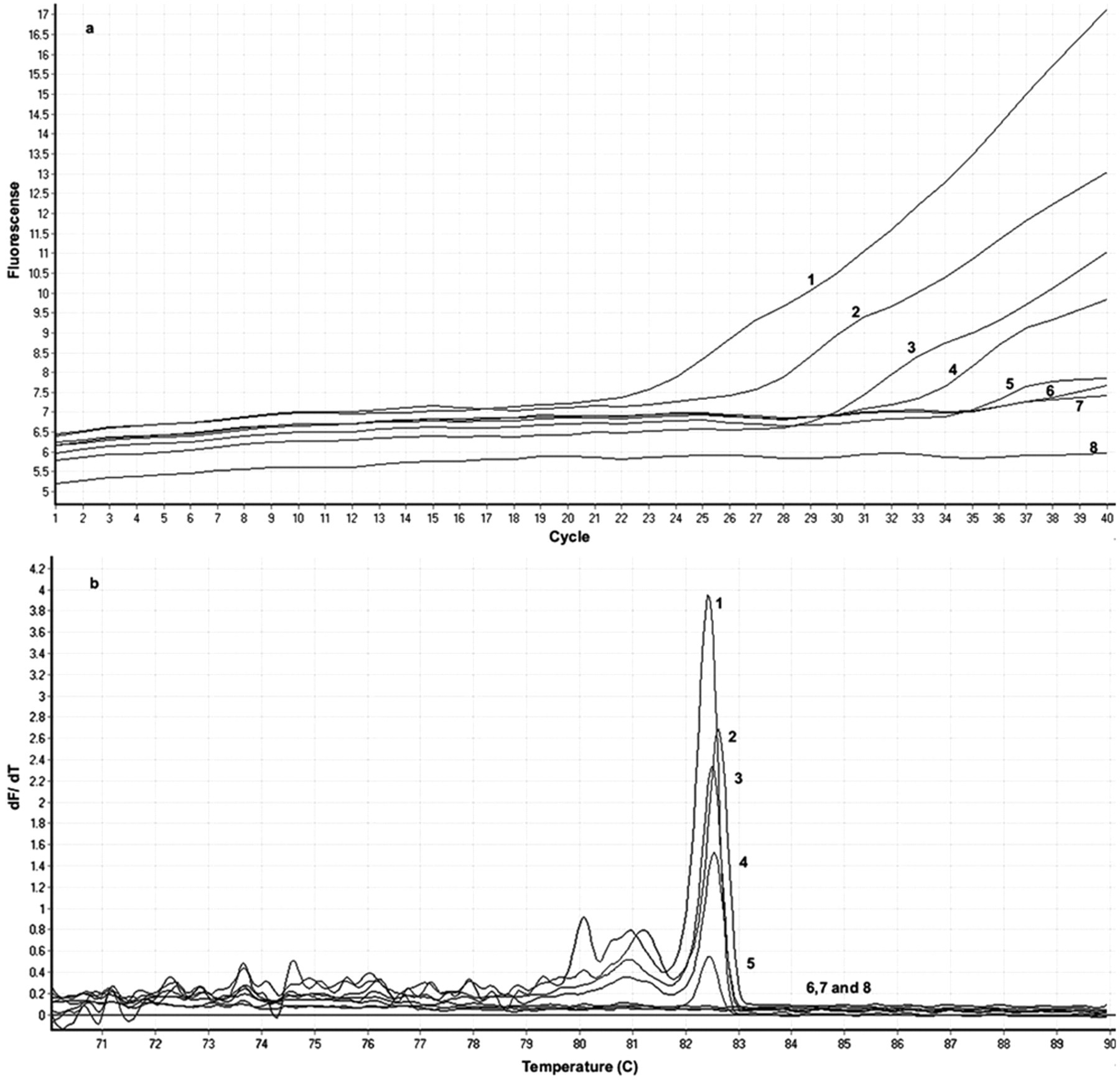
Representative (
Diagnostic sensitivity and specificity
Results of culture and real-time PCR for kidney samples corresponding to MAT are summarized in Table 1. Of 268 kidney samples, 17 were both culture and PCR positive. Two samples were culture negative but PCR positive and 3 were culture positive but PCR negative. The MAT results were available for 4 of those 5 samples, all of which were positive.
Number tested from each of 19 slaughter lines of farmed deer in New Zealand; number, kidney culture, real-time polymerase chain reaction (PCR), and microscopic agglutination test (MAT) positive; and the titer range for Leptospira serovars Hardjo-bovis and Pomona.*
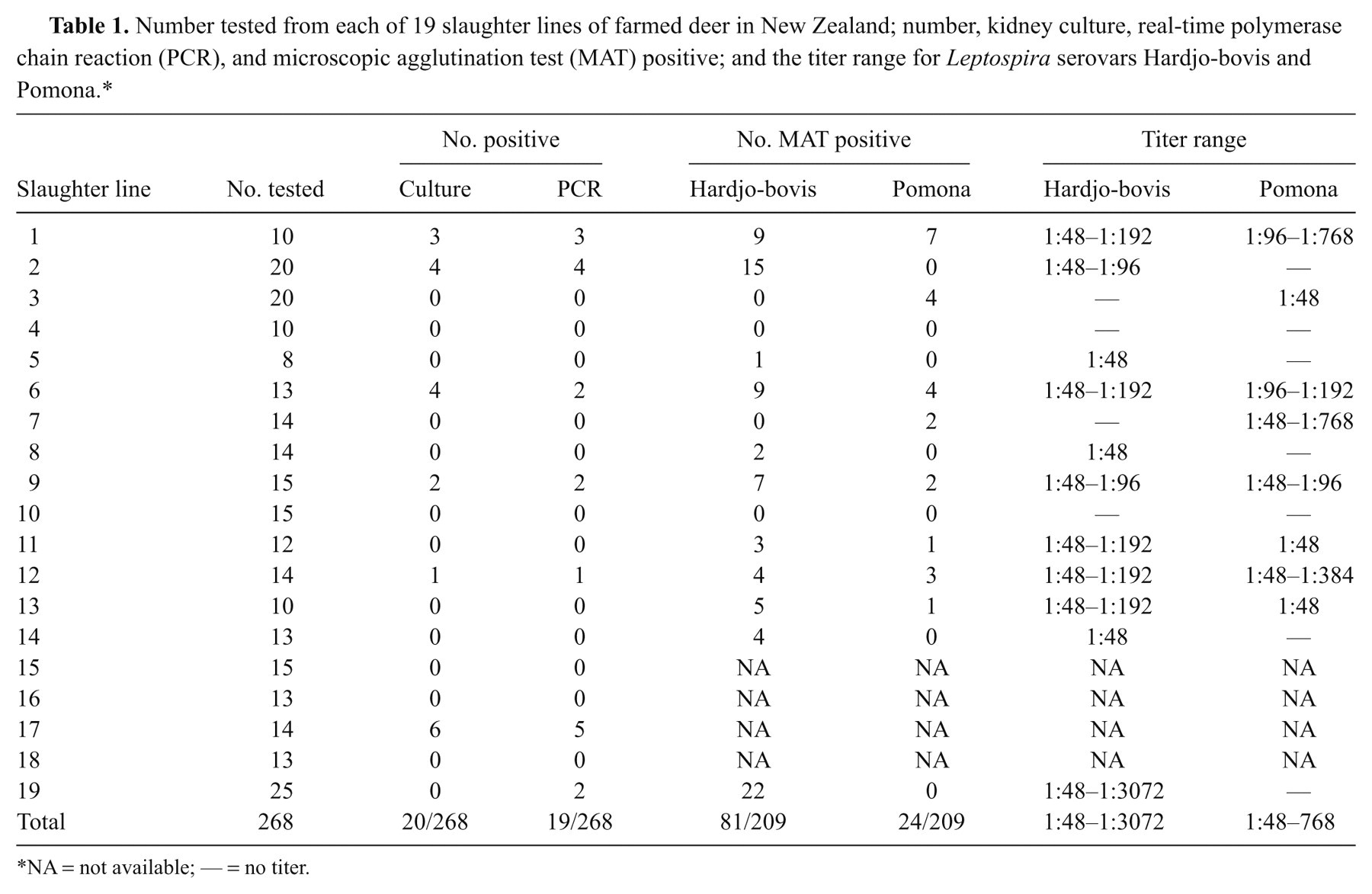
NA = not available; — = no titer.
Data of real-time PCR on kidney against culture and its performance are presented in Table 2. The sensitivity and specificity were 85% and 99.2%, respectively. The positive predictive value, negative predictive value, and kappa (agreement test) were 89.5%, 98.8%, and 86.2%, respectively. Of 11 real-time PCR-positive, fresh kidney samples, 7 (64%) were positive after being frozen at –20°C, and none were positive after being fixed in 10% neutral buffered formalin solution.
Number of kidney samples from 19 slaughter lines of farmed deer in New Zealand that were real-time polymerase chain reaction (PCR)- and culture-positive or -negative for Leptospira spp. and performance statistics of real-time PCR.*
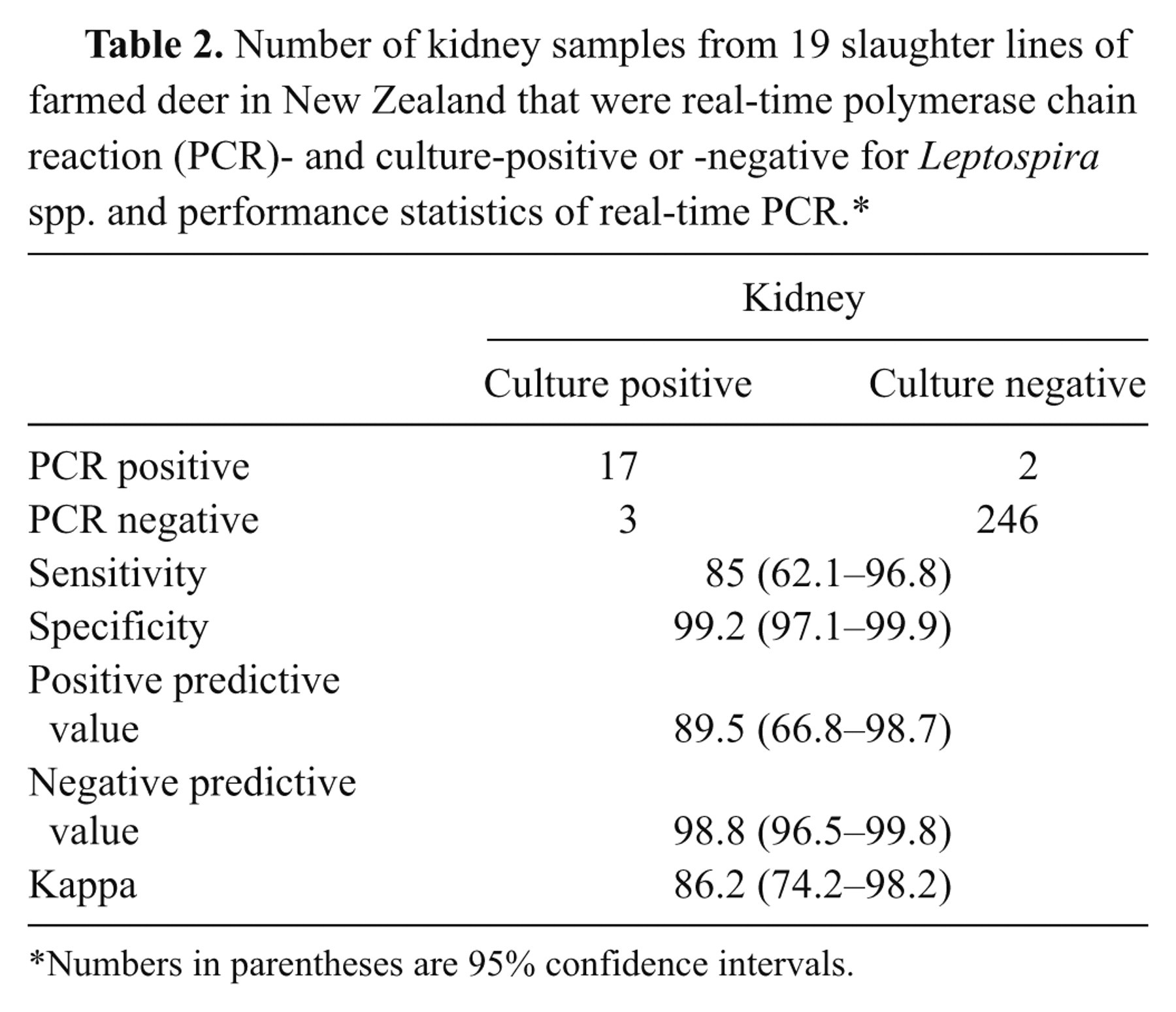
Numbers in parentheses are 95% confidence intervals.
Data of real-time PCR on urine against inoculated samples at the detection limit of 103 cells/ml and its performance are presented in Table 3. Of 30 inoculated urine samples, 29 were positive by real-time PCR. None of 83 Leptospira-free urine samples was positive by real-time PCR. The analytical sensitivity and specificity of the SYTO9 PCR assay in comparison to inoculated samples as the gold standard were 96.7% and 100%, respectively.
Number of Leptospira-free deer urine samples inoculated with field isolates of Leptospira serovar Hardjo-bovis; samples not inoculated, which were real-time polymerase chain reaction (PCR)-positive or -negative; and sensitivity and specificity of real-time PCR.*
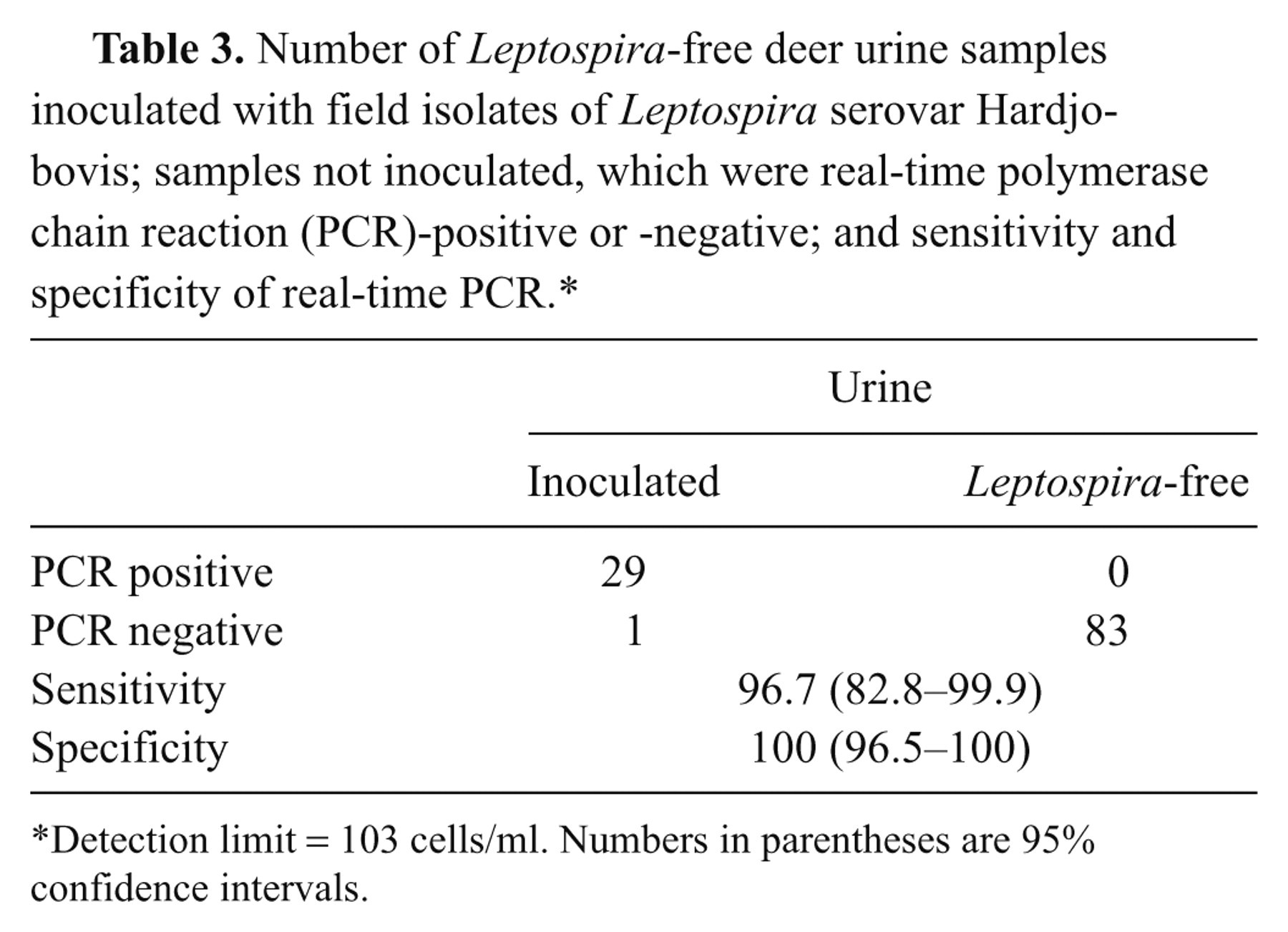
Detection limit = 103 cells/ml. Numbers in parentheses are 95% confidence intervals.
Quantification of urinary shedding
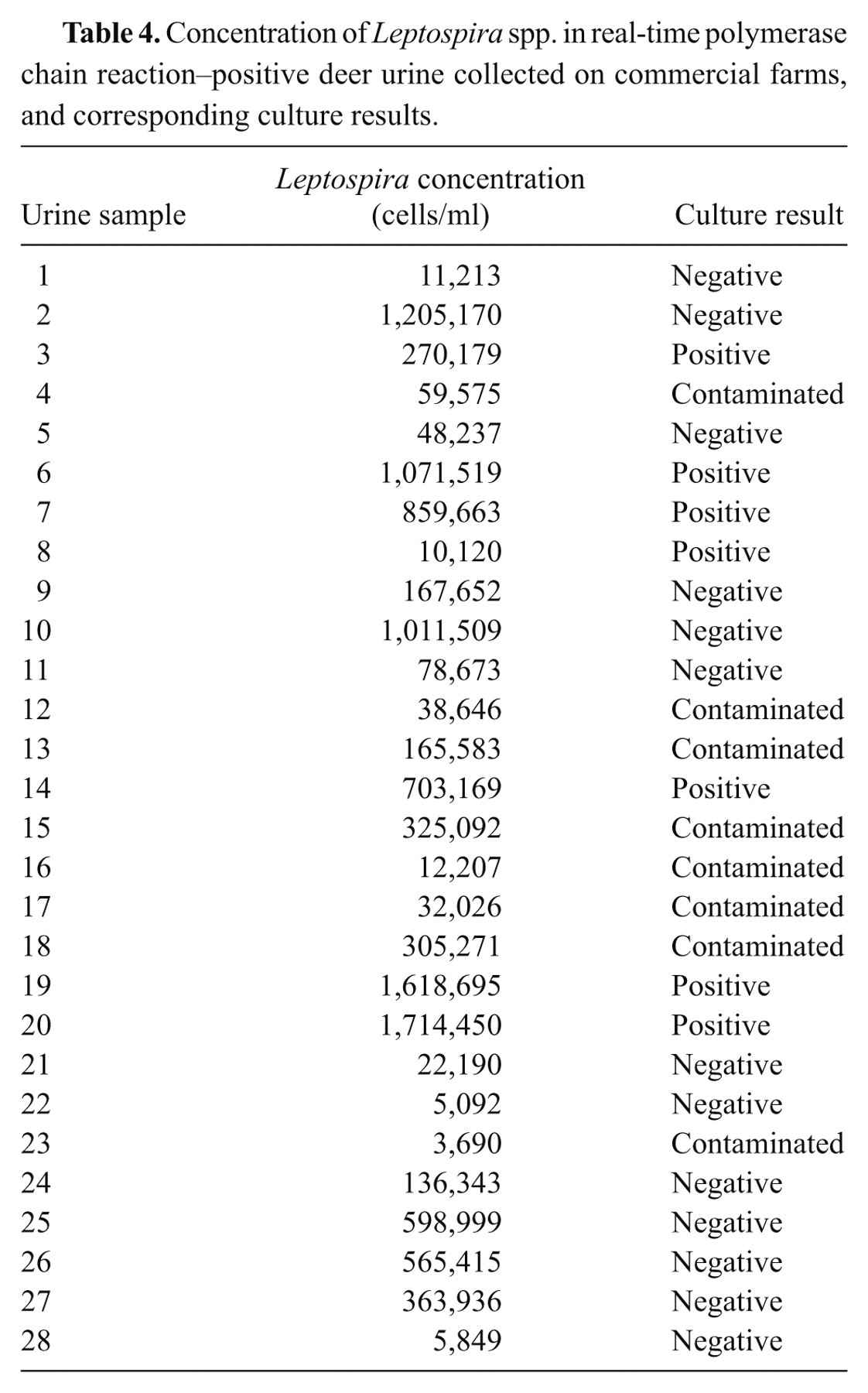
The concentration of leptospires from 28 real-time PCR-positive deer urine samples was between 3,690 and 1,714,450 cells/ml (Table 4). The lowest concentration of leptospires that was cultured from deer urine was 10,120 cells/ml (no culture result was available for sample 23, with a lower count, due to contamination). All 7 isolates derived from urine culture were serotyped as serovar Hardjo-bovis using standard antisera typing method.
Concentration of Leptospira spp. in real-time polymerase chain reaction–positive deer urine collected on commercial farms, and corresponding culture results.
Identification of pathogenic Leptospira species
The gyrB gene sequences of 14 unknown isolates were compared with those deposited on the NCBI database with BLAST search. Thirteen isolates were 100% homologous with the DNA sequence of L. borgpetersenii serovar Hardjo-bovis (NCBI accession nos. CP000350.1 and CP000348.1) and 1 isolate was 100% homologous with 3 different gyrB gene sequences of L. interrogans serovar Pomona (NCBI accession no. AY896738.1), L. interrogans serovar Medanensis (AY896746.1), and L. interrogans serovar Canicola (AY896745.1). There was 100% agreement between the gyrB gene sequencing and conventional antisera typing of 13 isolates being serovar Hardjo-bovis and 1 isolate being serovar Pomona. Phylogenetic analysis has confirmed 100% similarity between 13 isolates of serovar Hardjo-bovis that all differed from the single Pomona isolate. In addition, DNA sequences of 3 amplicons derived directly from deer kidney and 7 amplicons derived directly from deer urine were 100% homologous with the DNA sequence of L. borgpetersenii serovar Hardjo-bovis (NCBI accession nos. CP000350.1 and CP000348.1), when a BLAST search was performed.
Discussion
The current study evaluated and validated the performance of a diagnostic PCR assay for use on deer kidney tissue and urine as a research and diagnostic tool for determining infection, carrier, and shedding status of New Zealand farmed deer. A SYTO9 real-time PCR was developed using primers previously designed for a conserved region of gyrB sequences of pathogenic Leptospira species. 30 The study used real-time PCR directly on kidney and urine samples from farmed deer with evaluation against culture as the gold standard, and quantified pathogenic leptospires shed naturally in animal urine. The study also identified specific Leptospira species by DNA sequencing of PCR amplicons.
Based on a previously described method, 30 PCR conditions were optimized to suit the genetic analyzer. f The SYTO9 double-stranded DNA intercalating dye was adopted, as it has shown to have advantage over SYBR Green I 24 being easily adapted using standard conventional PCR reagents. That this assay does not need a specific probe like TaqMan or fluorescence resonance energy transfer makes it less expensive and able to run on a larger scale than other assays.
The lowest detection limit of the assay was determined by DNA extracted from known concentrations of leptospires measured in a Petroff–Hausser counting chamber and dark-field microscopy. The detection limit of the PCR using the DNA extracted from dilution of leptospires mixed with Leptospira-free deer urine was approximately 103 cells/ml or equivalent to 2–10 copies/reaction. This was consistent with another study that reported the detection limit of 103 cells/ml in inoculated human serum 27 and 3–10 copies/reaction in inoculated human serum and urine. 21
The assay showed 85% sensitivity (95% confidence interval [CI]: 62.1–96.8%), 99.2% specificity [95% CI: 97.1–99.9%]) and “excellent” agreement (kappa = 86.2%, 95% CI: 74.2–98.2%) with kidney culture as the gold standard. Failure of the PCR to detect leptospires in some culture-positive kidney samples may be due to leptospire concentrations below the detection limit. The DNA extracted from tissue can contain inhibitors interfering with PCR reactions, 15 so inhibitory substances may have been present. A further possible reason for 2 PCR-positive kidney samples being culture negative may be lack of viable organisms at the time of processing since those samples were from a DSP in Southland, a long distance from the laboratory, requiring more than 24 hr before processing. Samples from the other DSP were placed in culture within approximately 6 hr of collection. It is notable that the source animals for those 2 samples were seroreactive to serovar Hardjo-bovis with high titers consistent with recent exposure and/or current infection with leptospires. Hence, the authors assert that there is a high probability that the real-time PCR result does indicate infection.
Long-term storage of kidney samples for use in DNA extraction and real-time PCR was investigated on a limited number of specimens in the current study. The lowered sensitivity from frozen compared with fresh samples and the inability to detect DNA in formalin-fixed tissues suggest that kidney samples need to be processed fresh to achieve the highest sensitivity.
Approximately 30% of urine cultures from deer on farms were contaminated in culture whereas real-time PCR returned several positive results from such samples. Determination of the analytical sensitivity of real-time PCR was therefore undertaken using urine artificially inoculated with leptospires showing the lowest detection limit to be 103 cells/ml. The analytical sensitivity at this concentration and above was 96.7% (95% CI: 82.8–99.9%) and the specificity was 100% (95% CI: 96.5–100%). These results were comparable with a previous study, 29 which reported a diagnostic sensitivity of 96.4% and a specificity of 99.5% on patient sera using primer pairs Lepto F/Lepto R. Those primers, which were designed from the 16S ribosomal RNA gene, detected leptospiral DNA in both serum and seeded urine samples. 31
In addition to detection of the organism per se, the ability of real-time PCR to quantify the concentration of leptospires in clinical samples provides diagnostic benefits, for example, in evaluating antibiotic efficiency by post-treatment clearance of leptospires in urine. The technique may also be used for epidemiological studies involving Leptospira shedding and possibly even surface contamination when studying potential risk factors for disease transmission. One study has developed a real-time PCR assay to measure the concentration of leptospiral DNA in patient’s sera, reporting a range of 80–39,000 cells/ml, that informed the prognosis for patients suffering from leptospirosis. 23 To the authors’ knowledge, the current study is one of the first reporting detectable concentrations of Leptospira serovar Hardjo-bovis shed in field samples of deer urine, finding a range from 3.7 × 103 to 1.7 × 106 cells/ml. This defined the detection limit of the developed PCR assay at 103 cells/ml, although it is possible that deer may shed lower concentrations of leptospires in urine. The lowest concentration of leptospires (serovar Hardjo-bovis) that could be cultured from deer urine was 104 cells/ml, which was higher than that from a previous report of culture of 102 cells/ml in water and 103 cells/ml in bovine semen (serovar Hardjo-prajitno). 16 Considering that urine samples collected on-farm are prone to contamination, as shown in the present study (approximately 30% of samples were contaminated), the detection limit of urine culture is plausible and likely explains the higher sensitivity of real-time PCR over urine culture. However, urine from other sources (e.g., human beings) collected under aseptic conditions may yield a higher proportion of positive cultures.
A limitation of PCR-based diagnosis of leptospiral infection is the inability to identify the infecting leptospires at the species, serogroup, or serovar level. 22,23 The infecting serovar may be predicted by combining real-time PCR results with the MAT since data from the current study have shown that approximately 85% of kidneys positive to real-time PCR were from animals positive to the MAT. However, caveats to such conjecture are in those cases of early infection when the host immunity has not been activated, or in the event of dual or multiple serovar infections.
Species identification can be addressed by amplicon sequencing of PCR product of gyrB gene. 30 All 14 unknown isolates and 10 real-time PCR positive samples subjected to PCR amplification and sequencing of gyrB gene resulted in 100% homology with sequences deposited on the NCBI database when a BLAST search was performed. In addition, the BLAST search results matched fully with standard antisera typing showing that 13 isolates were identified as serovar Hardjo-bovis and 1 as serovar Pomona. The BLAST search results and phylogenetic analysis agreed with the previous findings 30 that sequencing of gyrB gene could differentiate pathogenic leptospires to species level but not to serovar level. However, since serovars Hardjo-bovis and Pomona are the most prevalent serovars in New Zealand livestock and belong to different species (L. borgpetersenii and L. interrogans), these techniques represent a rapid tool for identifying pathogenic Leptospira species and the likely serovar from clinical samples in New Zealand. Similar deduction would be appropriate in regions where only a few species and serovars of pathogenic leptospires are encountered. For more accurate genotypic classification, Multilocus Sequence Typing (MLST) is the most up-to-date method that has been developed and claimed to have high discriminatory power, reproducibility, and robustness. 1,32
In conclusion, a real-time PCR assay based on SYTO9 technology for the detection of pathogenic leptospires in kidney and urine samples of farmed deer in New Zealand has been validated. The assay was clinically evaluated against the gold standard (culture) and found to have comparable diagnostic sensitivity, specificity, and predictive values for kidney samples, and better sensitivity for urine samples. Such assay identifies pathogenic Leptospira species when combined with a DNA sequencing method. The method is simple, rapid, and easily adapted with conventional PCR reagents, which makes it less expensive than commercial real-time PCR reagents. It has potential for application as a research tool for the determination of carrier or shedding status of animal species for epidemiological studies and evaluation of vaccine efficacy, and as a clinical diagnostic tool on tissue and urine, and potentially blood samples, for rapid diagnosis in animals and human beings.
Footnotes
Acknowledgements
The authors would like to thank the participating deer farmers for access to deer samples, Venison Packers Feilding Ltd. and Alliance Group Makarewa Ltd., for their cooperation with animal sampling; Geoff Purchas; visiting students and post graduates within Massey University Deer Research Group for technical assistance; Lee Smythe of the WHO/FAO/OIE reference laboratory for leptospirosis in Brisbane, Australia and his team for advice on recent laboratory techniques for Leptospira; and Errol Kwan of the mEpiLab, Massey University for advice in molecular techniques.
a.
Vacutainer®, BD, Franklin Lakes, NJ.
b.
High Pure PCR Template Preparation Kit, Roche, Mannheim, Germany.
c.
Salix®, Intervet Ltd., Tauranga, New Zealand.
d.
Sigma-Aldrich, St Louis, MO.
e.
Invitrogen, Eugene, OR.
f.
Rotor-Gene 6000, Corbett Research, Mortlake, Australia.
g.
High Pure PCR Product Purification Kit, Roche, Mannheim, Germany.
h.
Perkin-Elmer Applied Biosystems, Norwalk, CT.
i.
Biomatters Ltd., Auckland, New Zealand.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The current project was funded by Intervet/Schering Plough Animal Health and Massey University.