
Abstract
The current study describes the development of a set of 5 multiplex polymerase chain reaction (mPCR) assays for the simultaneous detection of abortive infection agents in bovine fetal tissues, including Brucella spp., Leptospira spp., and Campylobacter fetus (mPCR1); Hammondia heydorni, Neospora caninum, and Toxoplasma gondii (mPCR2); Coxiella burnetii and Chlamydophila psittaci (mPCR3); Mycoplasma bovis, Mycoplasma bovigenitalium, and Ureaplasma diversum (mPCR4); and Bovine viral diarrhea virus (BVDV) and Bovine herpesvirus-1 (BoHV-1; mPCR5). The protocol was tested on different tissue samples collected from 50 aborted bovine fetuses, and it showed that out of the 50 fetuses, 7 (14%, mPCR2) were PCR-positive for N. caninum, 4 (8%, mPCR5) were PCR-positive for BVDV, and 2 (4%, mPCR4) were PCR-positive for U. diversum. The results obtained by using each multiplex PCR were 100% concordant with those obtained by using the respective PCR assays targeting single genes on the same specimens. Moreover, all multiplex PCR assays on clinical samples were compared with reference methods, obtaining a perfect accordance in all samples and confirming the validity of the set of multiplex PCR assays. The proposed set of multiplex PCR assays is, therefore, suitable for the simultaneous detection of the main infectious agents responsible for bovine abortion.
Introduction
Abortion among dairy cows is one of the major causes of economic losses in the cattle industry. Although the risk of abortion depends on several factors (i.e., genetic abnormalities, heat stress, toxic agents), infectious agents are likely to be one of the most important risk factors associated with abortions. 6,47 A variety of infectious agents have been reported to cause bovine abortion throughout the world. Bovine brucellosis and leptospirosis are distributed worldwide, and their importance is related not only with economic losses in animal production but also with a consistent risk for human health. Reproductive disorders, such as abortions and premature births, may be the only clinical signs of these bacterial diseases in pregnant cows. Coxiella burnetii, an obligate intracellular parasite with a worldwide distribution, is the causative agent of both acute and chronic Q fever in human beings as well as a cause of abortion and infertility in several mammalian species. 22–24 Although infection is often unapparent in cattle, there is increasing evidence that C. burnetii infection is associated with abortions and stillbirths. 2 Chlamydial infections may lead to overt clinical diseases, such as placental and fetal infections, with subsequent abortion, sterility, and, occasionally, mastitis. Frequently, this infection persists in animals as clinically unapparent, with shedding of the pathogen over long periods. 42 Campylobacter fetus may be found in the genital tract of cattle and sheep, in which it may cause genital tract infection and sporadic abortions. 21,39 Ureaplasma diversum and Mycoplasma bovigenitalium are generally thought to be relatively uncommon causes of abortions, although Ureaplasma can cause outbreak of abortions if it is introduced into a clean, previously unexposed herd. However, it is often difficult to determine if one of these agents is the cause of abortion since they can also be found in the reproductive tract of healthy cows. 7 Mycoplasma bovis has been associated with mastitis, infertility, endometritis, salpingitis, pneumonia, and arthritis as well as abortion. 35 Among viral abortive agents, Bovine viral diarrhea virus (BVDV) and Bovine herpesvirus-1 (BoHV-1) are of major concern in many countries, except in those where specific eradication programs have been successfully completed. Infection with BVDV is closely associated with a high incidence of diarrhea, respiratory disorders, and abortion, causing considerable economic loss. 19 The causative agent of infectious bovine rhinotracheitis (IBR), BoHV-1, is a DNA virus frequently associated with abortion in naturally and experimentally infected cattle. 26,28,32,36 Infection with BoHV-1 may cause other serious reproductive complications in artificially inseminated cows, such as infertility, endometritis, and embryonic absorption. 16,17,48 Virus isolation for the diagnosis of BVDV and BoHV-1 is a relatively low-sensitive test that is a laborious, costly, and time-consuming technique that sometimes takes 1–3 weeks to be completed.
Among parasites, apicomplexan protozoa represent an important cause of abortion. Even if Neospora caninum is considered to be the main agent involved in cattle abortions, 8 Toxoplasma gondii and Hammondia spp. have also been associated with abortion in ruminants. 1 Tissue cysts of the genera Hammondia, Toxoplasma, and Neospora are difficult to distinguish from each other histologically. 20,25 Therefore, diagnosis based on molecular assays can help to easily detect the specific organism involved.
Bacteriological isolation for the diagnosis of bovine brucellosis and leptospirosis is usually employed, but it is difficult, time consuming, hazardous, and sometimes inconclusive. 9,18,29 Routine diagnosis of Q fever is usually made by the use of serological tests, 33 which have the potential disadvantage of indicating post-exposure rather than persistence of infection. Diagnosis of chlamydial infections in animals still represents a considerable challenge. 42 Isolation in cell culture remains difficult, time consuming, and depends on the presence of sufficient numbers of viable bacteria. The reliability of standard diagnostic procedures for C. fetus, which are based on phenotypic methods, is difficult because of growth nutritional and atmospheric requirements and has been questioned. 30,31,40 Diagnosis of Mycoplasma infections relies on culture, which requires rapid transport of the clinical sample from the farm to the laboratory, the use of specialized media, and expertise in mycoplasma culture and identification.
Few polymerase chain reaction (PCR) protocols, with multiplex target genes, have been proposed to identify infectious agents. 4,37 Each method shows different features and needs specific annealing temperatures. No PCR protocols, which could assist in a more complete screening of these pathogens, are yet available. The present report proposes a set of multiplex (m)PCR assays to combine specific detection of 13 infectious agents responsible for bovine abortion, with substantial savings of time and costs compared to the current techniques used.
Materials and methods
Positive controls
For the present study, as positive controls for mPCR, genomic DNA extracted from the bacteria Brucella abortus (field strain Veterinary Medical Research Institute for Piemonte, Liguria and Valle D’Aosta [IZS] 1098), Chlamydophila psittaci (field strain IZS 100474-02), Leptospira interrogans serovar Pomona (field strain IZS 3112/2009), C. fetus (American Type Culture Collection [ATCC] 19438), M. bovis (field strain IZS 4511), Mycoplasma bovigenitalium (field strain IZS 3910), U. diversum (field strain Faculty of Veterinary Medicine of Turin [FMV] 475), C. burnetii Nine Mile phase I (ATCC VR-615), and from the parasites N. caninum (NC-1 strain), T. gondii (RH strain), Hammondia heydorni (HHA-GER1 strain) as well as RNA and DNA extracted from the viruses BVDV (strain Oregon C24 and field isolates) and BoHV-1 (strain Iowa and field isolates), respectively, were used.
Polymerase chain reaction assay
Different pairs of primers specific for each of the 13 infectious agent’s target genes were synthesized and selected from the literature or designed on the basis of alignment of published gene sequences (Table 1). In particular, each pair of primers was able to identify a highly conserved fragment in all infectious agents considered in the study. For example, the BVDV primer set was able to detect all ruminant pestiviruses so far described, including BVDV-1 and -2 and Border disease virus (BDV). All primer sequences were compared with one another, and a homology search was performed against the GenBank database for sequence similarity using the basic local alignment search tool (BLAST). a At first, in order to optimize the method, individual pair of primers were tested by PCR, using DNA or RNA prepared from positive controls. Amplicons were purified using a commercial purification kit b and sequenced to confirm the identity of the PCR-amplified products. The product sequencing was performed using PCR-derived primers in an automated DNA sequencer c by the dideoxy chain termination method with fluorescent dye terminators. The chromatograms of the nucleotide sequences were visualized using the software CHROMAS 2.0 d and submitted for BLAST analysis. a Sequences were then aligned using multiple-alignment software provided in the BioEdit package, version 7.0.5.2. 12
Development of a set of multiplex standard polymerase chain reaction (PCR) assays for the identification of infectious agents from aborted bovine clinical samples: simplex PCR assays used to assess presence of extracted DNA and RNA; the targeted genes and primers used for the detection of studied infectious agents; analytical sensitivity of multiplex PCR assays obtained from serial dilutions of plasmidic DNA.*
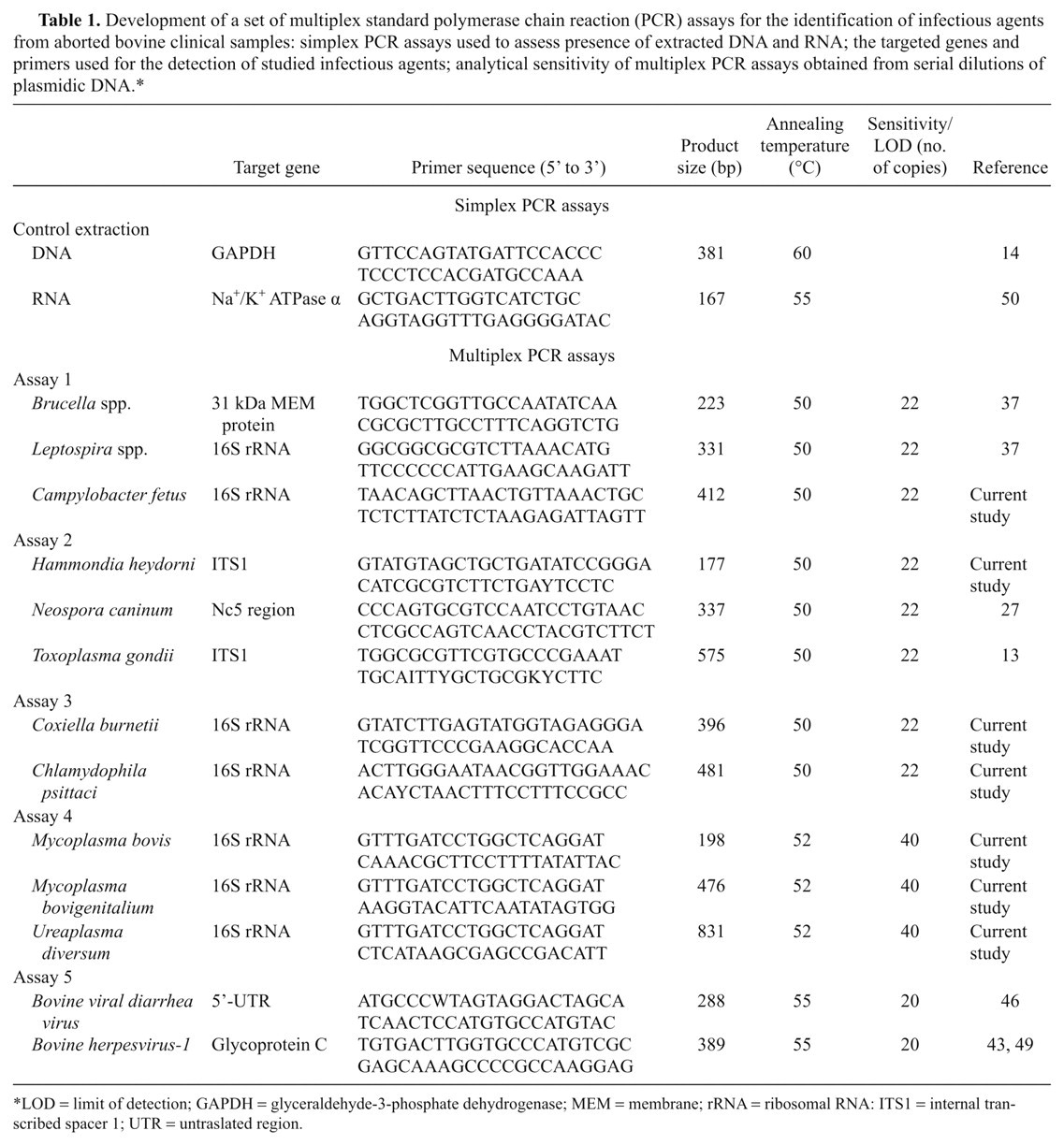
LOD = limit of detection; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; MEM = membrane; rRNA = ribosomal RNA: ITS1 = internal transcribed spacer 1; UTR = untraslated region.
Multiplex polymerase chain reaction assay conditions
The 5 mPCR assays were designed to simultaneously detect Brucella spp., Leptospira spp., and C. fetus (mPCR1); H. heydorni, N. caninum, and T. gondii (mPCR2); C. burnetii, C. psittaci (mPCR3); M. bovis, M. bovigenitalium, and U. diversum (mPCR4); and BVDV and BoHV-1 (mPCR5). Multiplex reactions were designed to be used in the same cycle conditions, and all selected primers were characterized as having annealing temperatures ranging from 50 to 55°C. The mPCR assays were performed in a total volume of 50 µl with a reaction mix containing 20 pmol of each primer e added to 5 µl of 10× reaction buffer, f 200 µmol of deoxyribonucleotide triphosphate (dNTP) e mix, and 0.5 µl of DNA polymerase. f Two µl of extracted DNA of each pathogen were added to the reaction mix. The thermal cycling protocol included an initial denaturation of 95°C for 15 min, followed by 40 cycles of 94°C for 1 min, annealing temperature for 1 min and extension of 72°C for 1 min. This was followed by a final extension step at 72°C for 10 min.
To detect BVDV and BoHV-1, a reverse transcription (RT)-PCR was carried out using an RT-PCR kit. g The reaction mixture was prepared as follows: RNase-free water, 10 µl of 5× RT-PCR buffer, 200 µmol of dNTP e mix, 15 pmol of each primer, e and 4 µl of RT-PCR enzyme mix. Two µl of extracted nucleic acid were added to the reaction mixture for a final volume of 50 µl per tube. The reaction cycles were set as follows: reverse transcription for 30 min at 50°C, initial PCR activation step for 15 min at 95°C, denaturation for 1 min at 94°C, annealing for 1 min at 55°C, extension for 1 min at 72°C, for 40 cycles, and final extension for 10 min at 72°C.
The PCR products were electrophoresed on 2% (w/v) agarose gel. e Gels were stained in 0.6 µg/ml ethidium bromide solution for 20 min. Products were visualized using an ultraviolet transilluminator, h and molecular weight sizes were determined by comparison with a 100–base pair (bp) DNA ladder plus. i
Multiplex polymerase chain reaction assay analytical sensitivity
The amplicons obtained were ligated to the pDrive cloning vector. j The ligated products were transformed into Escherichia coli, strain pMos, k and plated on lysogeny broth agar e containing ampicillin l (50 µg/ml), overlaid with isopropyl β-D-1-thiogalactopyranoside e (0.1 M) and bromo-chloro-indolyl-galactopyranoside e (X-gal; 50 mg/ml). The plates were incubated at 37°C overnight and screened for blue and/or white colonies. The white recombinant colonies were picked up and inoculated in 3 ml of lysogeny broth e containing ampicillin. The plasmids were isolated from an overnight culture using a commercial method, m and screening was performed by digesting the recombinant plasmid with EcoR1 to release the insert. Plasmid DNA was quantified using a fluorimetric method, and a 10-fold dilution series of the obtained plasmids was used to evaluate the analytical sensitivity of the PCR panel.
Multiplex polymerase chain reaction assay specificity
The performance of each individual pair of primers in the PCR assay was evaluated using DNA and RNA prepared from the positive controls and from DNA and RNA of 11 non-target microorganisms (Salmonella enteritidis, Yersinia enterocolitica, E. coli, Staphylococcus aureus, Staphylococcus epidermidis, Pasteurella multocida, Proteus spp., Streptococcus spp., Pseudomonas aeruginosa, Borrelia burgdorferi, and Helicobacter pylori).
Application of multiplex polymerase chain reaction assays on clinical samples
Clinical samples from 50 aborted bovine fetuses without lesions suggestive of a specific infectious disease were randomly selected from different farms under the regional control program for bovine abortion in Piedmont (northwest Italy). Abomasal contents and a set of organs (including lung, spleen, liver, kidney, brain, and muscle) were collected and refrigerated in an RNA stabilization reagent n for 24 hr and subsequently stored at −20°C until used.
Nucleic acid extraction
To extract both DNA and RNA, a protocol designed for the isolation of total nucleic acids from the samples collected was adopted by using a commercial purification kit o in combination with a homogenizer p and a workstation, q according to the manufacturer’s instructions. In brief, 300 µl of abomasal content and brain, along with 10 mg of spleen, lung, liver, kidney, and muscle were separately homogenized with 300 µl of RNeasy lysis buffer on the homogenizer for 2 min at 30 Hz. In this protocol, the DNase treatment step was omitted to allow purification of high-quality total nucleic acids (DNA and RNA) in high yields. The elution volume was set at 50 µl in the study. Concentrations of the obtained nucleic acids were determined using a spectrophotometer. r Successful nucleic acid extraction was verified for all tissues of 50 fetuses examined, using 2 different single PCR assays (Table 1): the first, for DNA extraction control, specific for bovine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) DNA sequence, 13 and the second, for RNA extraction control, using RT-PCR to amplify the sodium and/or potassium ATPase α subunit, enabled to differentiate by size the genomic DNA (approximately 2 kb) from complementary DNA (167 bp). 50 Amplified products of extraction controls were analyzed on agarose gel electrophoresis as a preliminary step before all assays.
Multiplex polymerase chain reaction assays
The set of 5 different multiplex assays as well as each of the simplex assays were then performed on 5 µl of nucleic acids extracted from a selected set of fetal tissues (namely brain, abomasal content, and spleen). In cases of positive results, the presence of target sequence was also evaluated in lung, muscle, kidney, and liver of the same fetus. To confirm the specificity of PCR products, all amplicons were purified b and sequenced. c
Standard reference methods
For those assays in which validation was not available from published literature, such as an in-house developed PCR assay, results obtained from clinical samples were compared with standard methods, including bacteriological isolation for Mycoplasma, Ureaplasma, and Campylobacter and PCR analysis for C. psittaci, C. burnetii, and H. heydorni. 5,38,41
Results
Multiplex polymerase chain reaction assays
Under carefully controlled simplex PCR conditions, optimized on positive control nucleic acids, amplicons of the expected size were obtained with all primer pairs. Sequence analysis of respective PCR products showed significantly high degree similarity (ranging from 99% to perfect match, related to the origin of positive control: field vs. reference strains) with the corresponding sequences from the GenBank database.
Based on the obtained results by matching the selected primer pairs in different ways, 5 mPCR assays correctly amplified all target genes (Fig. 1). Each primer pair amplified DNA fragments of the predicted size, and amplifications were specific for the corresponding gene. When primer pairs were combined in each multiplex assay, no primers interfered with the amplification of other targets, and, therefore, no false negatives were observed. Details of the nucleotide sequence, specific reference, and predicted length of resulting amplicon for each primer pair are listed in Table 1. Analytical sensitivity/limit of detection (LOD) of the PCR panel obtained from serial dilutions of plasmid DNA are shown in Table 1. Based on nucleic acid concentration of a subset of infectious agents available in purity, the detection limit reached less than 1 fg.
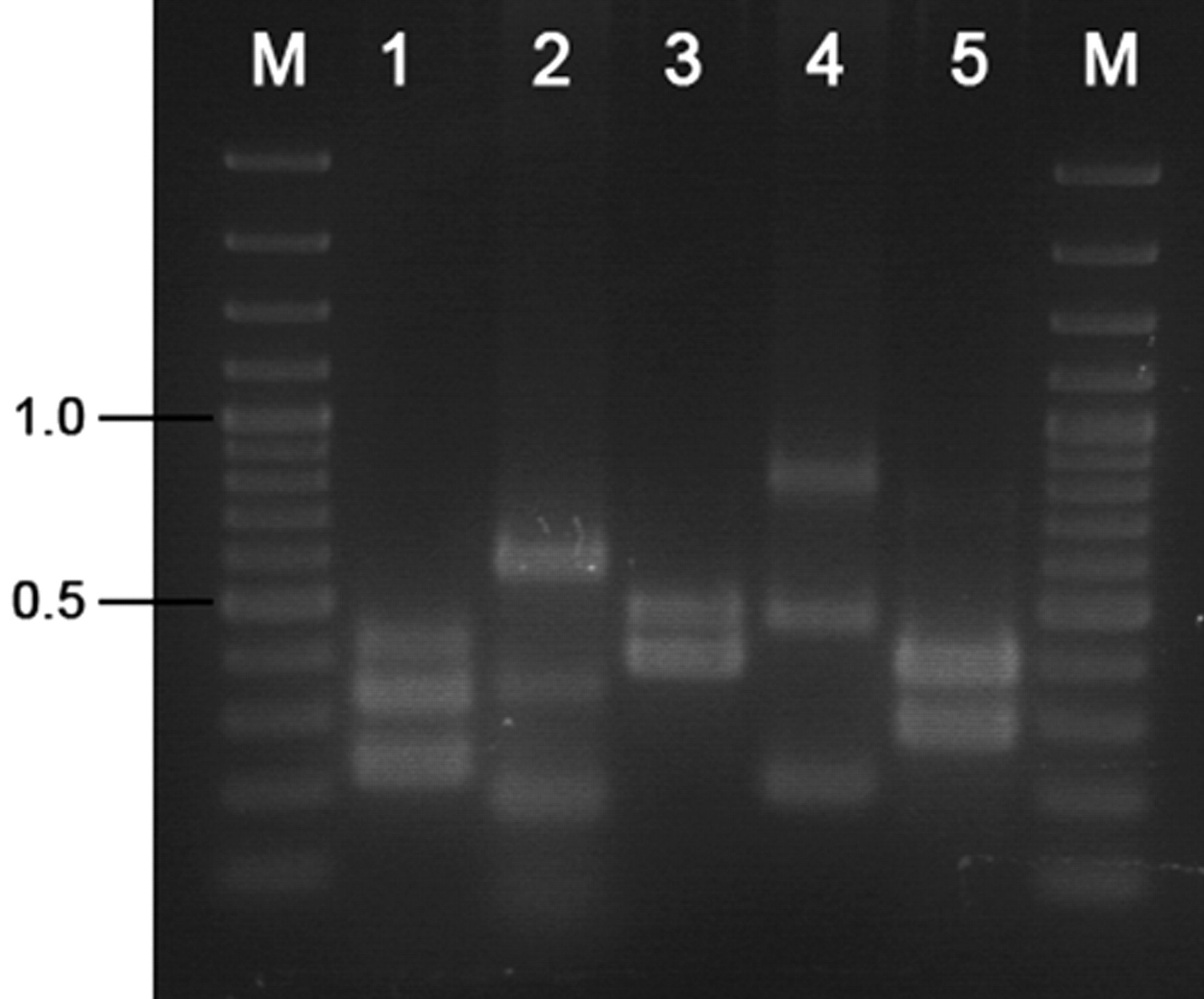
Amplification products obtained by the set of multiplex polymerase chain reaction (mPCR) assays. Lane M: 100–base pair DNA ladder plus; lane 1: mPCR1 (Brucella spp., Leptospira spp., and Campylobacter fetus); lane 2: mPCR2 (Hammondia heydorni, Neospora caninum, and Toxoplasma gondii); lane 3: mPCR3 (Coxiella burnetii and Chlamydophila psittaci); lane 4: mPCR4 (Mycoplasma bovis, Mycoplasma bovigenitalium, and Ureaplasma diversum); lane 5: mPCR5 (Bovine viral diarrhea virus and Bovine herpesvirus-1).
Application of multiplex polymerase chain reaction assays on clinical samples
The concentrations of extracted nucleic acids measured by spectrophotometry ranged from 500 ng/µl to 800 ng/µl. Amplification of the 381–base pair (bp) fragment from the GAPDH gene and of the 167-bp products from the sodium and/or potassium ATPase α subunit, respectively, confirmed successful DNA and RNA extraction from all types of tissues extracted (abomasal content, brain, lung, spleen, liver, kidney, and muscle), confirming the validity of extraction method used. Of the 50 fetuses examined, 7 (14%, mPCR2) were PCR-positive for N. caninum, 4 (8%, mPCR5) or BVDV, and 2 (4%, mPCR4) for U. diversum. In detail, the 7 positive fetus brains for N. caninum, were also positive in kidney (n = 3), in muscle (n = 2), and in abomasal content (n = 2); PCR-positive results for U. diversum were detected in lung and abomasal contents of 2 different fetuses. The 4 BVDV-positive cases yielded positive results both in abomasal samples and in all fetal tissues examined. Co-infection with N. caninum and BVDV was recorded in 1 case. Figure 2 shows the results of mPCR assays on representative bovine fetal tissues examined. There was 100% agreement between the results obtained by mPCR assays and those obtained single PCR assays for the various agents studied. Sequencing of PCR products showed a similarity of 98–100% with the sequences of N. caninum (EF202080), BVDV (EU180032), and U. diversum (D78650) retrieved from the GenBank database. Moreover all in-house PCR-positive results on clinical samples were compared with reference methods obtaining a perfect accordance in all samples and confirming the validity of the set of mPCR assays.
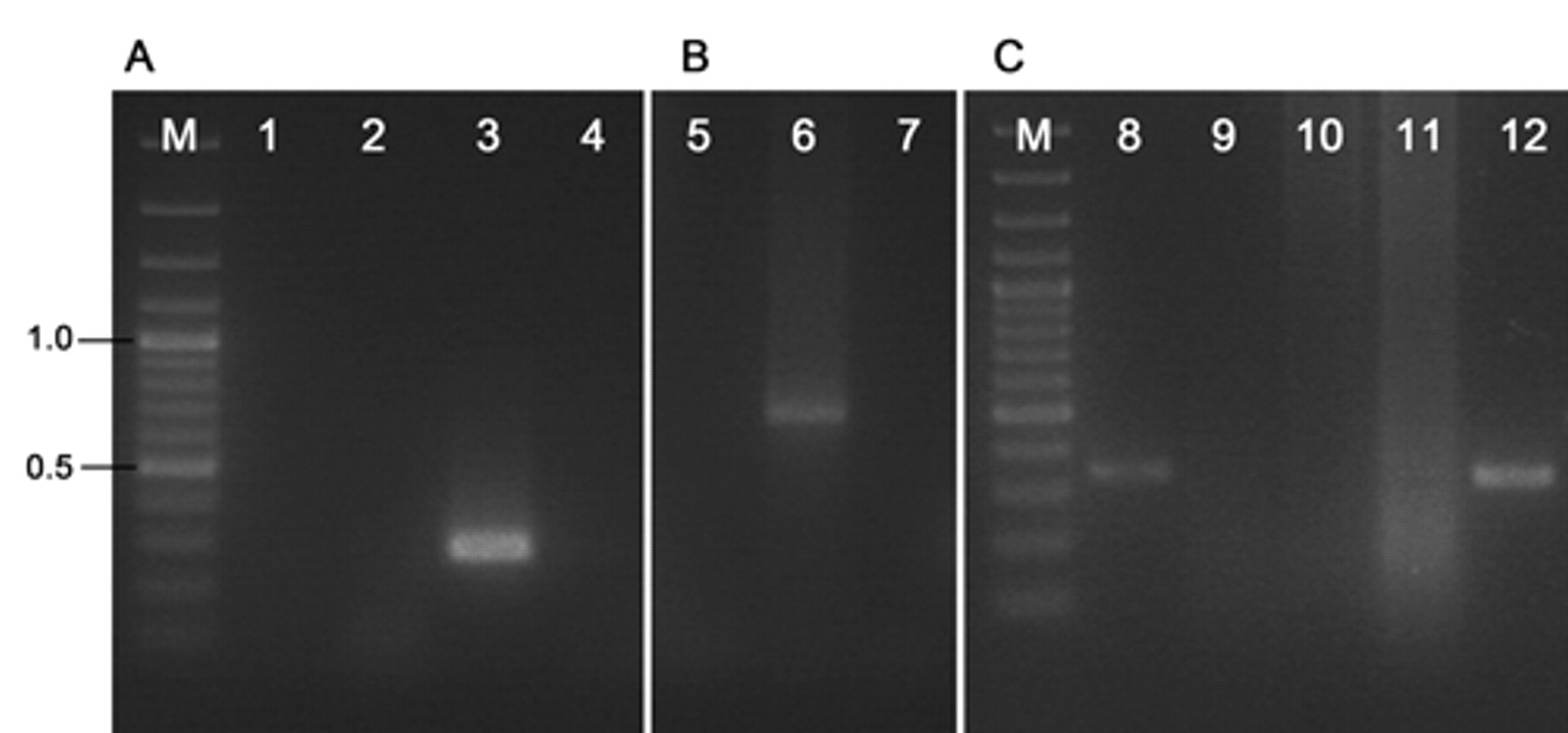
Multiplex polymerase chain reaction (mPCR) assays on representative bovine fetal tissues.
Discussion
In most cases, traditional approaches are not reliable enough to identify the infectious agents that cause bovine abortion, as they are often inconclusive and time consuming. Identification of some pathogens often requires the use of techniques such as assays on tissue culture, oligonucleotide probes, and fluorescent antibody tests, which are not usually part of the diagnostic laboratory routine. Although some PCR assays, both simplex and multiplex, have been previously described for detection of the 13 agents targeted in the current study, 3,15,44,45 there is, to the authors’ knowledge, no PCR panel for the simultaneous detection of all 13 agents in specimens from aborted bovine fetuses.
The results obtained in the present study highlight the presence of N. caninum, U. diversum, and BVDV in aborted bovine fetuses. In particular, N. caninum is associated to the occurrence of abortion in Piedmont cattle farms as found by others 11 who reported N. caninum from 34% of aborted fetuses in the same area. The findings in the present study confirm previous studies showing that the brain is the most effective tissue for detecting N. caninum in fetuses, even if N. caninum was also found in skeletal muscle and in kidney. 10,34 Four fetuses were PCR-positive for BVDV, suggesting that such virus plays a potential contributing role as an important abortive agent in the current territory, despite the extensive vaccination programs practiced as the main control measures. Co-infection with N. caninum and BVDV had previously been reported 8 : N. caninum–induced abortion outbreaks could be enhanced by the immunosuppressive effect of BVDV, which would increase the susceptibility to other infectious agents. The presence of U. diversum in fetal samples may not be directly associated with abortion but may be correlated with genital infections of pregnant cows. Indeed, Mycoplasma spp. are usually isolated from the bovine urogenital tract of healthy cows. 7 The high number of negative samples may be correlated not only with the low incidence of infections in Piedmont, such as brucellosis, which has been eradicated since 2008 (Commission Decision 2009/342/EC of 23 April 2009), but also with the decision to submit fetal samples from various farms to test randomly and not from livestock known to be infected with a particular infectious agent.
That 2 or more target sequences present in different amount in the same fetal tissue may cause false negative results regarding to the less prevalent agent, due to competitive PCR component, cannot be excluded. Such problem can potentially affect all multiplex targeting molecular assays. However, the data obtained using the set of multiplex PCR assays were in perfect accordance with those observed by the same PCR assays run in simplex, demonstrating the high reliability and sensitivity of the proposed panel. Further studies with a larger number of samples would be necessary to confirm whether these pathogens are consistent factors related to abortion in cattle in Piedmont.
In conclusion, the set of mPCR assays described in the present study would be a cost-effective way of simultaneously detecting the 13 leading infectious agents associated with bovine abortion in the Piedmont region. Application of this panel of mPCR assays is simpler, less expensive, and faster than the use of 13 single PCR assays.
Footnotes
a.
BLAST®, National Center for Biotechnology Information, Bethesda, MD.
b.
QIAquick PCR Purification Kit, Qiagen GmbH, Hilden, Germany.
c.
ABI Prism® 310 Genetic Analyser, Applied Biosystems, Milan, Italy.
d.
CHROMAS 2.0, Technelysium, Helensvale, Australia.
e.
Sigma-Aldrich, St. Louis, MO.
f.
HotStarTaq DNA polymerase, Qiagen GmbH, Hilden, Germany.
g.
OneStep RT-PCR Kit, Qiagen GmbH, Hilden, Germany.
h.
Gel Doc 2000, Bio-Rad Laboratories s.r.l, Milano, Italy.
i.
GeneRuler 100 bp DNA Ladder plus, Fermentas, M-Medical, Milano, Italy.
j.
PCR Cloning Kit, Qiagen GmbH, Hilden, Germany.
k.
GE Healthcare Europe GmbH, Milano, Italy.
l.
Pfizer Italia s.r.l., Rome, Italy.
m.
Miniprep Kit, Qiagen GmbH, Hilden, Germany.
n.
RNAlater RNA Stabilization Reagent, Qiagen GmbH, Hilden, Germany.
o.
EZ1 RNA Tissue Mini Kit, Qiagen GmbH, Hilden, Germany.
p.
Tissue Lyser Homogenizer, Qiagen GmbH, Hilden, Germany.
q.
BioRobot® EZ1 Workstation, Qiagen GmbH, Hilden, Germany.
r.
NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific Inc., Waltham, MA.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The current study was supported by a grant of Regione Piemonte, Italy (Funds: Ricerca Sanitaria Finalizzata, 2005).