
Abstract
Porcine chlamydial infection is an enzootic infectious disease caused by multiple members of the family Chlamydiaceae (e.g. Chlamydophila abortus, Chlamydia suis, and Chlamydophila pneumoniae). Rapid and accurate differentiation of these pathogens is critical in the control and prevention of disease. The aim of the current study was to develop a nested multiplex polymerase chain reaction (nmPCR) assay to simultaneously detect the 3 chlamydial pathogens in clinical samples. In the first round of the nmPCR, 1 pair of family-specific primers were used to amplify the 1,100 base pair (bp) fragment of chlamydial ompA gene. In the second round of the nmPCR, 4 inner primers were designed for Ch. abortus, C. suis, and Ch. pneumoniae. Each pathogen produced a specific amplicon with a size of 340 bp, 526 bp, and 267 bp respectively. The assay was sensitive and specific for detecting target pathogens in both cell cultures and clinical specimens. The results, incorporated with the improved rapid DNA extraction protocol, suggest that the nmPCR could be a promising assay for differential identification of different chlamydial strains in pigs.
Introduction
Chlamydial infection is considered as an enzootic infectious disease of pigs. 4,16 Concurrent infection of chlamydial pathogens with other pig diseases generally causes wasting, respiratory signs, diarrhea, enlargement of lymph nodes, and increased mortality. 31 The causative agents of porcine chlamydial disease include Chlamydophila abortus, Chlamydia suis, and Chlamydophila pneumoniae. 5,33 Chlamydophila abortus has not only been associated with enzootic abortion and other clinical signs in sheep and pigs, 21,26 but has also been related to cases of human disease. 22 Chlamydia suis, which is a close relative of Chlamydia trachomatis, has been found in semen and genital tracts of boars, sows, bulls, and bucks; the clinical signs of the infected animals varied from conjunctivitis, to enteritis, to pneumonia. 14,25 Although there is no evidence about the possibility of infection in human beings with C. suis, the emergence of removable tetracycline-resistant plasmids in C. suis suggests a potential threat for the food safety of human beings. 34 Although Ch. pneumoniae is considered a human infectious agent, evidence indicates that a wide range of hosts for Ch. pneumoniae, including human beings, koalas, horses, reptiles, and pigs, exists and that a possible reason for such a wide range of hosts may be that Ch. pneumoniae potentially crosses the various host barriers through the processes of gene decay and plasmid loss in such hosts. 3,23 The high prevalence of Ch. pneumoniae–specific antibodies (22.5%) in the sera of boars from breeding stations also seemed to be a potential health hazard for human beings. 5
Chlamydial infections can be detected either by isolation of pathogens or by screening for antibodies in tissue scrapings and smears, tissue sections, serum, and bodily secretions. 27,29 Because of the insufficient sensitivity, low specificity, and low pathogen loads of clinical samples, it is difficult to detect infection status by means of such tests. 29 Alternative methodologies based on nucleic acid amplification, such as polymerase chain reaction (PCR), which possess both unsurpassed sensitivity and specificity, have been recently used to diagnose human chlamydiosis. 2,24,30 Although real-time PCR assay and microarray-based methods are superior to traditional PCR assays in their sensitivity as well as throughput, the traditional PCR assays are easier to perform and standardize in different facilities. 28,29 The traditional PCR assays, which involve tedious DNA extraction procedures or combining a few pair of primers to detect 1 or very few targets in 1 reaction, are not convenient enough for the rapid identification of porcine chlamydial infection. 7 For example, the successful detection of all related Chlamydophila and Chlamydia spp. of veterinary importance needs 6 separate, species-specific real-time PCR assays. 20 For the diagnosis of enzootic abortion, a multiplex PCR for the differential detection of Ch. abortus and Chlamydophila pecorum and a duplex real-time PCR for the specific detection of Chlamydophila psittaci and Ch. abortus have been developed. 19 To the authors’ knowledge, a PCR assay for the simultaneous detection of the Ch. abortus, C. suis, and Ch. pneumoniae has not been applied. In the current study, a nested multiplex polymerase chain reaction (nmPCR) assay was developed to simultaneously detect such zooanthroponotic chlamydial pathogens from clinical samples.
Materials and methods
Reagents, cell, organisms, and their propagation
The following reagents were used: Eagle minimal essential medium (EMEM), a fetal calf serum (FBS), b genomic DNA (gDNA) isolation kit, c PCR reagents (including 10× PCR buffer, deoxyribonucleotide triphosphate [dNTP] mix, and Taq polymerase), d fluorescein isothiocyanate (FITC)-conjugated Chlamydiaceae-specific antibody, e and antibiotics (gentamicin, vancomycin, cycloheximide). f Chlamydophila abortus, C. suis, Ch. pneumoniae, and C. trachomatis were cultured in Henrietta Lacks (HeLa) 229 cell line as described in the World Organization for Animal Health (OIE) manual, with the following modifications. 35 Briefly, 100 µl of stock cultures were inoculated onto 24-well plates of exponentially growing HeLa 229 cell monolayers in EMEM supplemented with 5% FBS and antibiotics (gentamicin and vancomycin), and then centrifuged at 500 × g for 60 min at room temperature. The cultures were incubated for 3–5 days in a CO2 incubator following addition of 0.5 µg/ml of cycloheximide. The supernatant of the cell culture was harvested and mixed with 2× sucrose–phosphate–glutamate (SPG) buffer (149.2 g/l of sucrose, 1.02 g/l of monopotassium phosphate, 2.474 g/l of dipotassium phosphate, 1.442 g/l of L-glutamic acid). The elementary body contained in the supernatant was titrated by immunofluorescent staining with an FITC-conjugated Chlamydiaceae-specific antibody. The number of inclusions was counted in 20 random fields and calculated as inclusion-forming units (IFU) per milliliter. The supernatant of the cell culture was used for the PCR assay. Other organisms (see Table 1), including Pasteurella multocida, Legionella pneumophila, Mycoplasma hyopneumoniae, Brucella abortus, Listeria monocytogenes, Actinobacillus pleuropneumoniae, enterotoxigenic Escherichia coli, Haemophilus parasuis, and Klebsiella pneumoniae, were cultured in proper medium and used for DNA extraction.
Chlamydial strains used in the current study for the development of the nested multiplex polymerase chain reaction assay.*
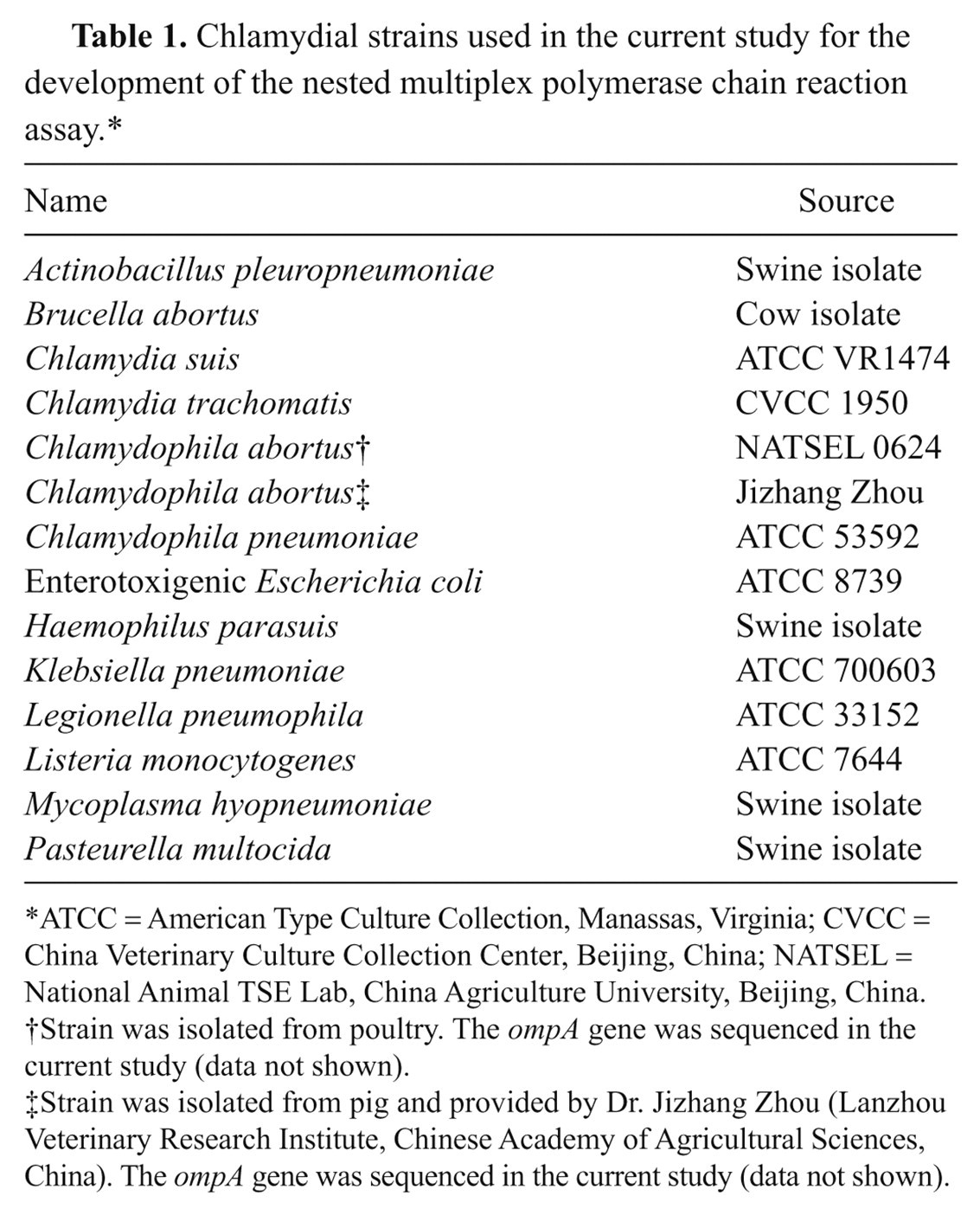
ATCC = American Type Culture Collection, Manassas, Virginia; CVCC = China Veterinary Culture Collection Center, Beijing, China; NATSEL = National Animal TSE Lab, China Agriculture University, Beijing, China.
Strain was isolated from poultry. The ompA gene was sequenced in the current study (data not shown).
Strain was isolated from pig and provided by Dr. Jizhang Zhou (Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China). The ompA gene was sequenced in the current study (data not shown).
DNA extraction and optimized protocol for swabs
The DNA was extracted from the organism cultures and clinical samples by using 2 different protocols. In protocol 1, DNA was extracted by using a silicon column–based gDNA isolation kit, c according to the manufacturer’s instructions. In protocol 2, samples were centrifuged at 500 × g for 5 min at room temperature, and the supernatant was collected. After centrifuging the supernatant at 10,000 × g for 30 min, the sediment was resuspended in 11 µl of Mini-Q water and the mixture boiled at 100°C for 15 min. Then, the mixture was centrifuged at 500 × g for 1 min, and all of the supernatant was used for the PCR assay.
To evaluate the reliability of protocol 2 for DNA extraction from swabs, approximately 10 ml of supernatants from chlamydia-free swabs were pooled by centrifuging at 500 × g for 5 min and stored at 4°C until use. A cell culture containing Ch. abortus (5 × 102 IFU/ml), C. suis (3 × 102 IFU/ml), and Ch. pneumoniae (2 × 102 IFU/ml) was mixed in equal volumes and serially diluted to 10−4 with the pooled supernatants. Using protocols 1 and 2 separately, the DNA was extracted from 300 µl of supernatant obtained at dilution, and then used for PCR assays.
Primer design
A multi-alignment–based criterion was used for primer design. 13 Primers were designed in the domains that were conserved among family or species, forming a nested strategy (Table 2). The primers were evaluated with the National Center for Biotechnology Information Basic Local Alignment Search Tool (BLAST) for their specificity, then synthesized. g
Primers used in the current study to amplify the chlamydial strains.*

R = A/G; Y = C/T; W = A/T; bp = base pair. Numbers in parentheses are length of product.
“→” indicates the direction is from upstream to downstream on the sense strand of the genomic DNA; “←” indicates the reverse direction.
Nested multiplex polymerase chain reaction assay
Polymerase chain reaction assay mixtures were prepared in a PCR workstation, and the amplification and analysis of PCR products were performed in separate locations. The PCR assay was optimized by varying primer concentrations and by methodical variation of each test parameter under standard PCR conditions. During the first round of the PCR assay, samples were amplified in a 25-µl reaction containing 1× PCR buffer, 200 µmol/l of dNTP mix, 1.5 mmol/l of MgCl2, 1.25 U of Taq polymerase, 0.3 µmol/l of Hup primer, 0.3 µmol/l of Hlow primer and 50–100 ng of DNA as the template. The thermal profile consisted of 1 cycle at 95°C for 5 min, followed by 40 cycles at 95°C for 30 sec, 59°C for 35 sec, and 72°C for 1 min, and 1 cycle of final extension at 72°C for 8 min. In the second round of the PCR assay, samples were amplified in a 25-µl reaction containing 1× PCR buffer, 200 µmol/l of dNTP mix, 2.0 mmol/l of MgCl2, 1 U of Taq polymerase, 0.4 µmol/l of Hup-n primer, 0.3 µmol/l of Slow primer, 0.2 µmol/l of Clow primer, 0.1 µmol/l of Flow primer, and 0.5–1.0 µl of product of the first round of nmPCR assay as the template. The thermal profile consisted of 1 cycle at 95°C for 5 min followed by 30 cycles at 95°C for 30 sec, 52°C for 30 sec, and 72°C for 30 sec, and 1 cycle of final extension at 72°C for 8 min.
The PCR products were electrophoresed on a 2% agarose gel stained with ethidium bromide and visualized under ultraviolet light. The product size of the first amplification round was 1,100 bp. The specific product size of the second round nmPCR assay was 340 bp (Ch. abortus), 526 bp (C. suis), and 267 bp (Ch. pneumoniae).
The evaluation of specificity and sensitivity
The DNA used in the specificity assay was extracted from a panel of pathogens listed in Table 1. First, the DNA was amplified by the first round nmPCR. Then, 1 µl of product from the first round PCR was amplified by the second round species-specific nmPCR. To assess the sensitivity, the supernatant of cell culture containing Ch. abortus (5 × 103 IFU/ml), C. suis (3 × 103 IFU/ml), and Ch. pneumoniae (2 × 103 IFU/ml) was mixed in equal volume and serially diluted to 10−4 with 0.1 mol/l of phosphate buffered saline. Then, using protocol 2, 300 µl of supernatant from each dilution was extracted, and DNA was amplified by nmPCR assay. Equal amounts of DNA were also amplified by the 16S ribosomal DNA (rDNA) nmPCR assay 18 and the 23S rDNA real-time PCR assay. 11
Evaluation of the nested multiplex polymerase chain reaction assay with clinical samples
Swabs (vaginal, nasopharyngeal, and rectal) were collected from 122 adult sows from 2 local breeding farms diagnosed with a high prevalence of anti-Chlamydiaceae antibody and subclinical reproductive and respiratory disease. Swabs were freshly soaked into 1× SPG buffer (supplemented with 10% fetal bovine serum, 500 µg/ml of streptomycin, 50 µg/ml of gentamicin, and 50 µg/ml of amphotericin B) and kept at 4°C. According to protocol 2, 300 µl of supernatant was used to extract DNA, and the DNA templates were detected by both nmPCR and 23S rDNA real-time PCR assays. Both negative (water) and positive controls (plasmid) were also included in the PCR assays.
Pathogen isolation and sequencing
Three hundred microliters of the swab supernatant was used for pathogen isolation, which was performed according to the OIE manual by inoculating the yolk sac of 6–7-day-old embryonated chicken eggs. After 7 days, the yolk sac and allantoic fluid were collected. Pathogens were examined by nmPCR assay. The negative samples were treated as described earlier and inoculated into chicken egg embryos for another passage. 20 The positive PCR products of the nmPCR assay were cloned into pMD19-T vector. Positive clones were sequenced and multi-aligned with the appropriate reference sequences (C. suis AF269274; Ch. pneumoniae M69230.1; Ch. abortus AF269256). The sequences were analyzed using Molecular Evolutionary Genetics Analysis (MEGA 4.0) software. 32
Results
Sequence analysis and primer design
A phylogenetic tree was used for the primer design by multi-alignment of sequences, which were grouped into 9 separate branches, with patterns identical to the 16S rDNA phylogenetic tree built by the international classification organization. 10 One pair of family-specific primers was designed on the family-specific domain and named Hup and Hlow. Another forward family-specific sense primer was designed 200 bp downstream of the primer Hup and named Hup-n. Three antisense primers specific for Ch. abortus, C. suis, and Ch. pneumoniae were individually designed on the species-specific domains that were located between Hup and Hlow primers. These primers formed the nested pattern (Fig. 1).
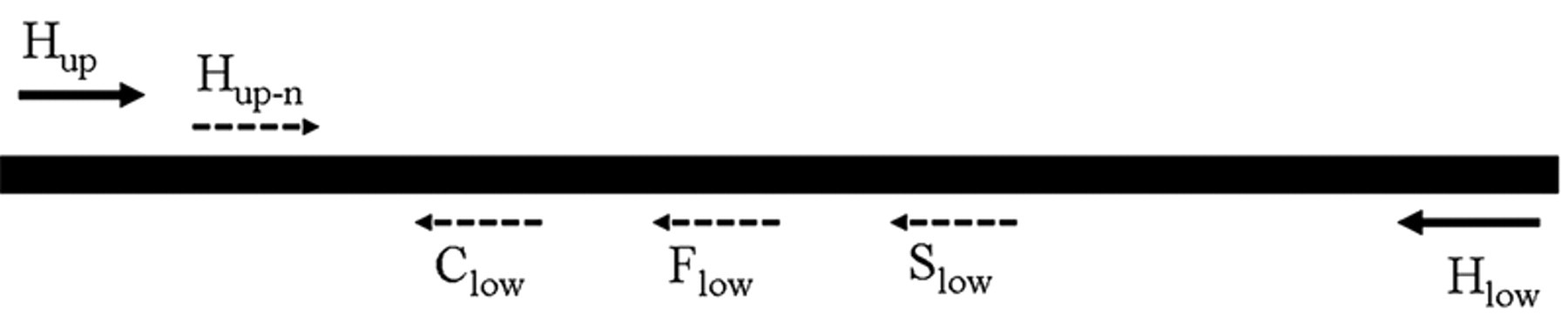
Primers used in the current study were spanning the ompA gene. Primers Hup, Hlow, and Hup-n were chlamydial family-specific. Primers Slow, Clow, and Flow were specific for Chlamydophila abortus, Chlamydophila pneumoniae, and Chlamydia suis, respectively.
Specificity of the nested multiplex polymerase chain reaction assays
The 1,100-bp family-specific fragment was amplified from Ch. abortus, C. suis, Ch. pneumonia, and C. trachomatis in the first round of nmPCR. There was no amplification from the other microorganisms (Fig. 2A). A second round of nmPCR further analyzed the products of the first round amplification. As shown in Figure 2B, fragments specific to Ch. abortus, C. suis, and Ch. pneumoniae were successfully amplified, respectively; however, C. trachomatis and other microorganisms could not yield specific amplicons. The DNA of Ch. abortus, C. suis, and Ch. pneumoniae were examined by PCR assays separately and as mixtures. The PCR assay could simultaneously detect these 3 species in every arrangement as shown in Figure 3.
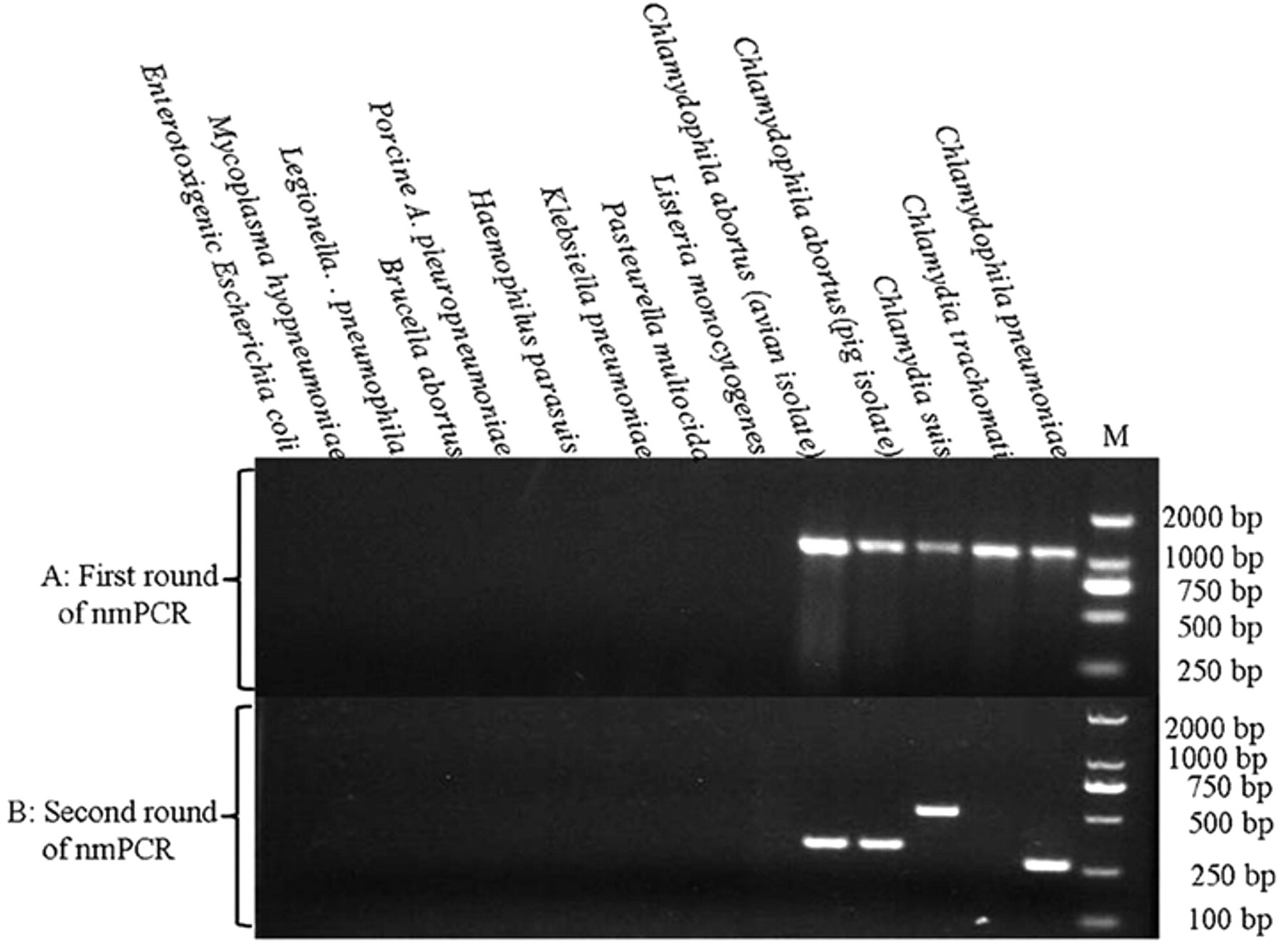
Results of the specificity test of the nested multiplex polymerase chain reaction (nmPCR) assay.
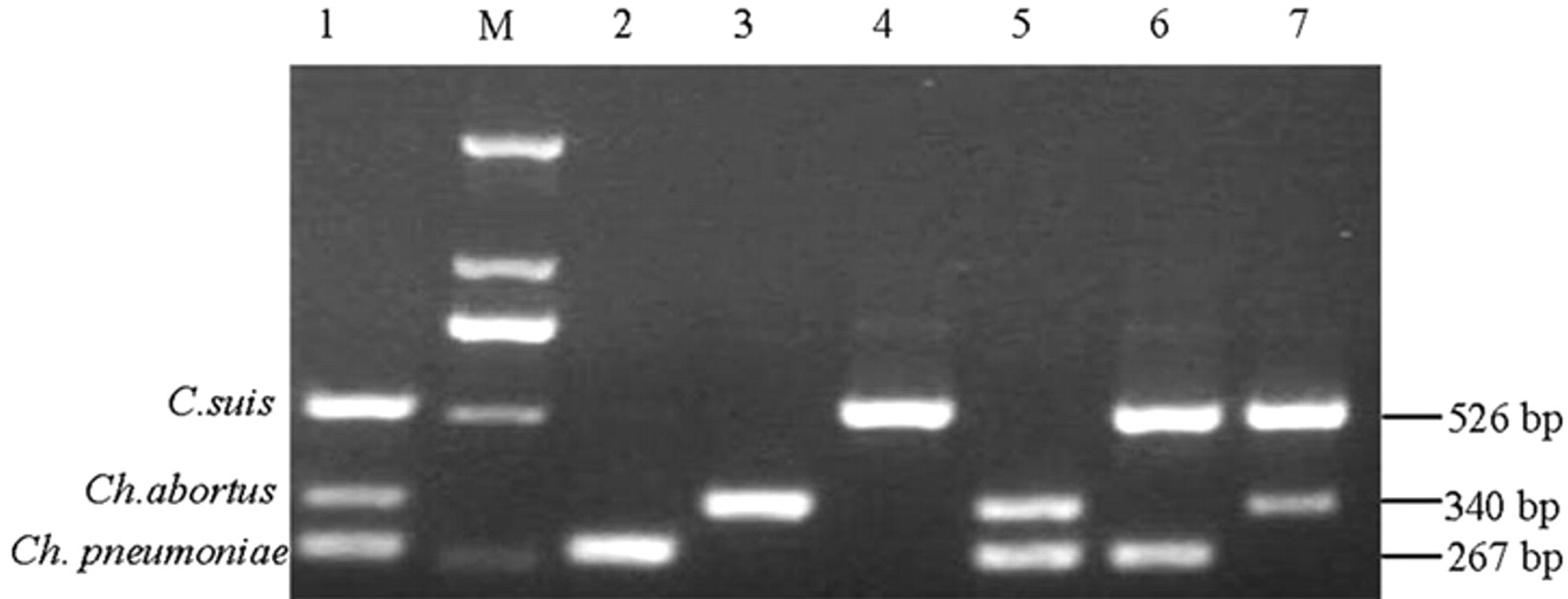
Simultaneous detection of multiple chlamydial organisms in 1 reaction. Lane 1: Chlamydophila abortus+ , Chlamydia suis+ , and Chlamydophila pneumonia+ ; lane M: DL2000 DNA marker; lane 2: Ch. pneumoniae+ ; lane 3: Ch. abortus+ ; lane 4: C. suis+ ; lane 5: Ch. pneumoniae + and Ch. abortus+ ; lane 6: Ch. pneumoniae+ and C. suis+ ; lane 7: Ch. abortus+ and C. suis+ .
Sensitivity of the nested multiplex polymerase chain reaction assays
As shown in Figure 4, nmPCR assay could simultaneously detect Ch. abortus, C. suis, and Ch. pneumoniae at dilutions as low as 10−3. The sensitivity of the 2-step nmPCR assay developed was under 5 IFU/ml, and it was 10 times more sensitive than that of the 16S rDNA nPCR and 23S rDNA real-time PCR arrays as shown in Figure 5.
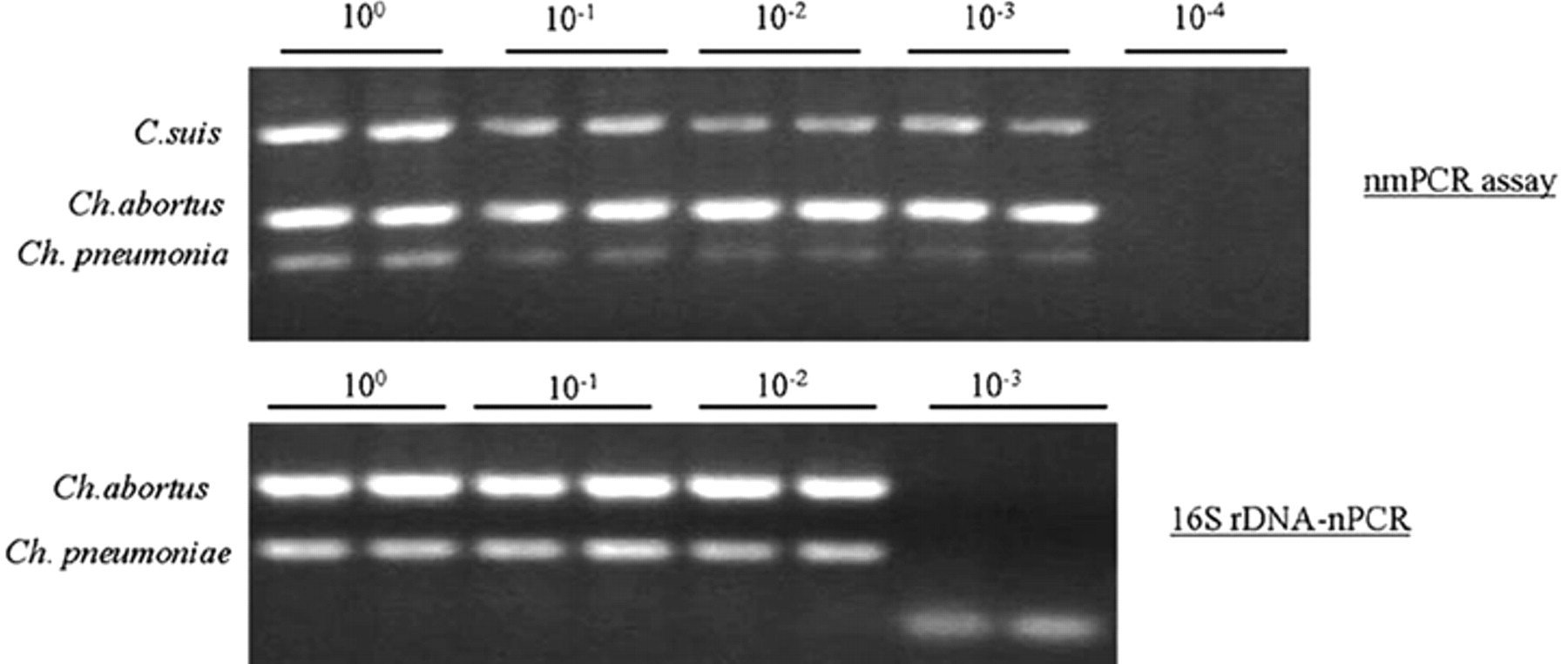
Comparison of the analytical sensitivity between 2 nested multiplex polymerase chain reaction assays.
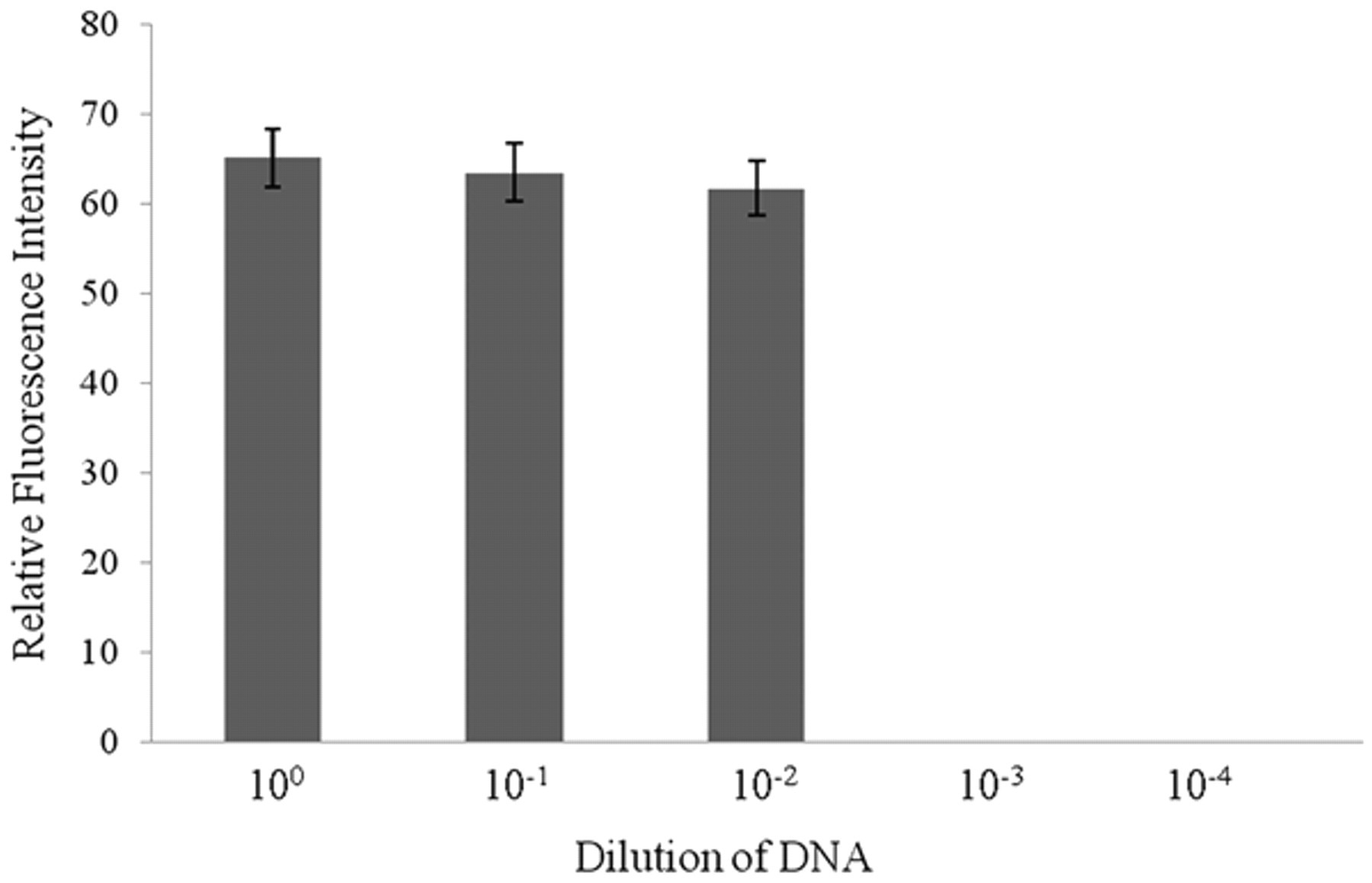
Results of the sensitivity test of the 23S real-time polymerase chain reaction (PCR) assay. The serially diluted chlamydial organisms were plotted with relative fluorescence intensity. Relative fluorescence intensity was calculated by: (100 – threshold Ct) × 100%, where Ct indicates the threshold cycle numbers in real-time PCR. The relative fluorescence intensity of the negative samples was considered as zero.
Optimized protocol for swab DNA extraction
The 2 protocols could extract sufficient DNA for successful amplification of specific fragments of Ch. abortus, C. suis, and Ch. pneumoniae at the 10−1dilution. At the dilution of 10−2, the nmPCR of DNA extracted by protocol 1 (commercial DNA kit) could not amplify visible fragments. However, there were visible amplifications of all the 3 targets by protocol 2 (Fig. 6). Thus, protocol 2 was 10 times more efficient than protocol 1 for DNA extraction.
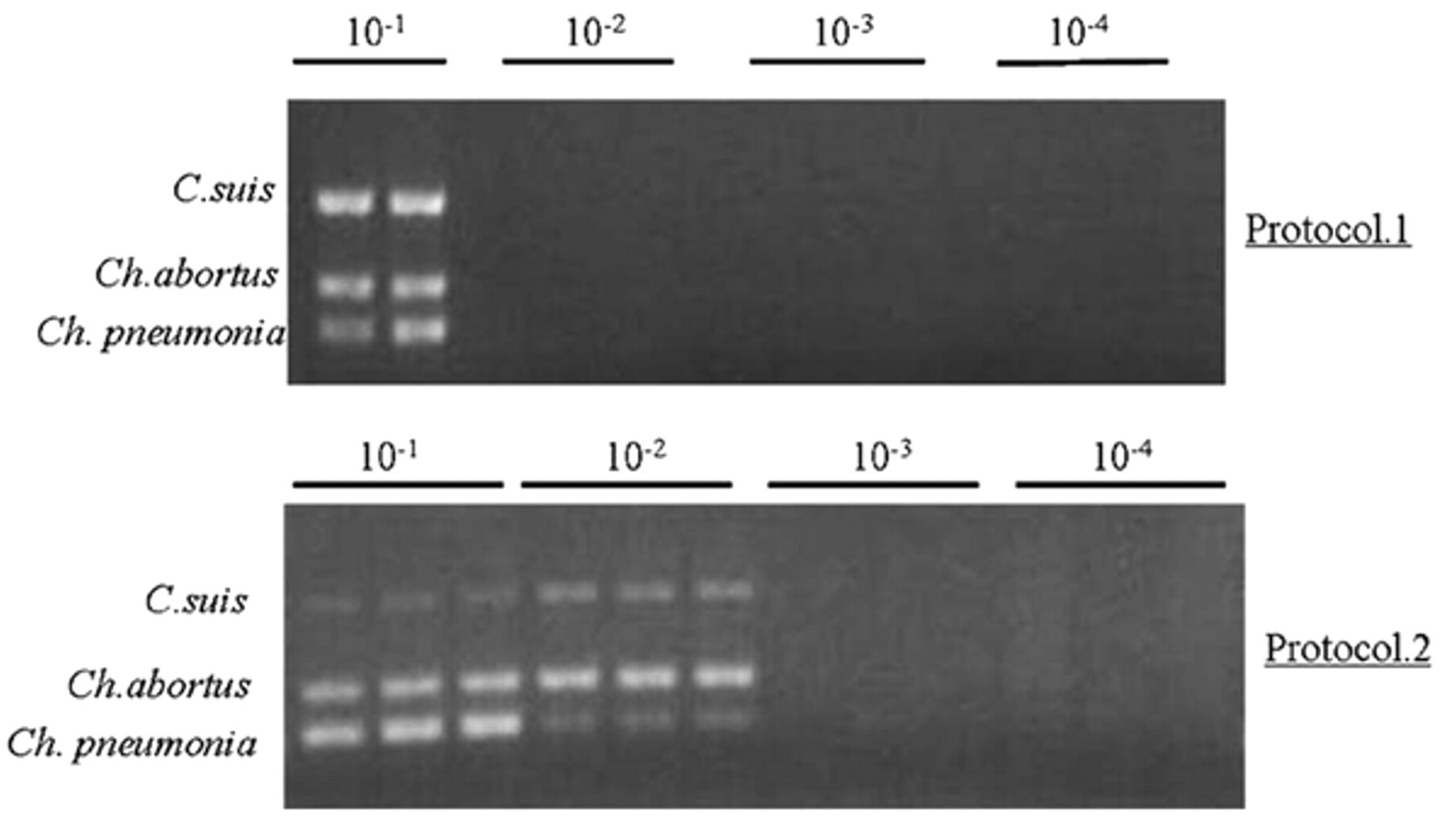
Optimized protocols used for the DNA extraction of swabs to detect Chlamydophila abortus, Chlamydia suis, and Chlamydophila pneumonia.
Evaluation of the nested multiplex polymerase chain reaction assay with clinical samples
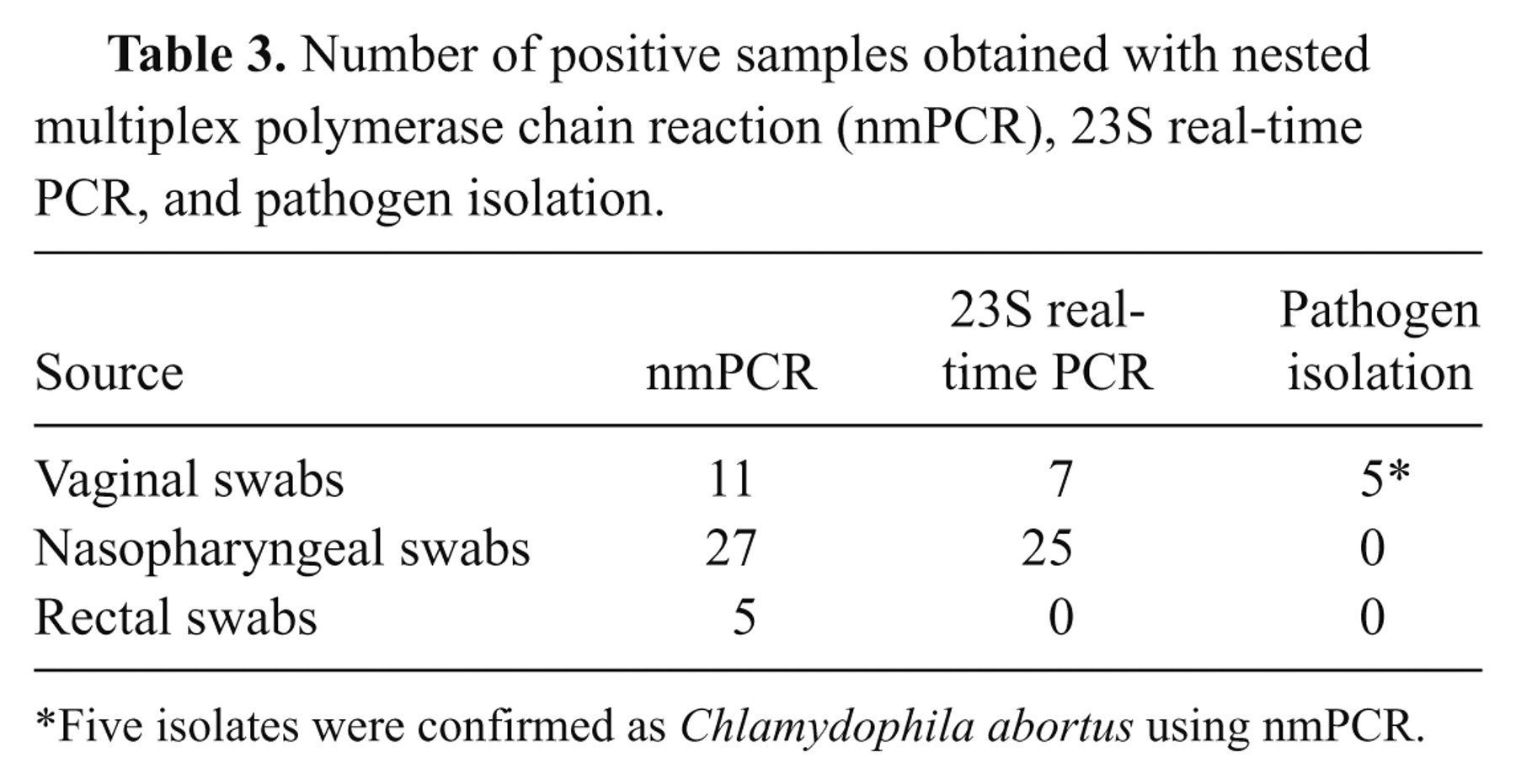
A different prevalence of chlamydial infection was observed in swabs by different methods (Table 3). The positive rate of the nmPCR assay is 9% in vaginal swabs, 3% and 5% higher than that of 23S real-time PCR and pathogen isolation, respectively. The nmPCR assay gave rise to 22% positive rate in nasopharyngeal swabs, which is 2% higher than that of 23S real-time PCR. While the positive rate resulted by the nmPCR assay is 4% in rectal swabs, it was negative from pathogen isolation and 23S real-time PCR. The infection type of the sow detected by nmPCR assay was shown in Table 4. Chlamydophila abortus was only detected from vaginal swabs. Six vaginal swabs and 3 rectal swabs were positive for C. suis. Meanwhile, Ch. pneumoniae was found in nasopharyngeal and rectal swabs. However, the 23S real-time PCR could not differentiate among them.
Number of positive samples obtained with nested multiplex polymerase chain reaction (nmPCR), 23S real-time PCR, and pathogen isolation.
Five isolates were confirmed as Chlamydophila abortus using nmPCR.
Number of positive samples detected by nested multiplex polymerase chain reaction assay.

Positive products of the second round nmPCR were cloned into pMD19-T vector, and 16 clones were successfully sequenced. The multi-alignment shown illustrates that all C. suis clones were obviously different from the reference strain (C. suis S45 strain) maintained in the authors’ laboratory (Fig. 7). The sequences of Ch. pneumoniae and Ch. abortus acquired from nmPCR products were approximately the same as the reference strains.
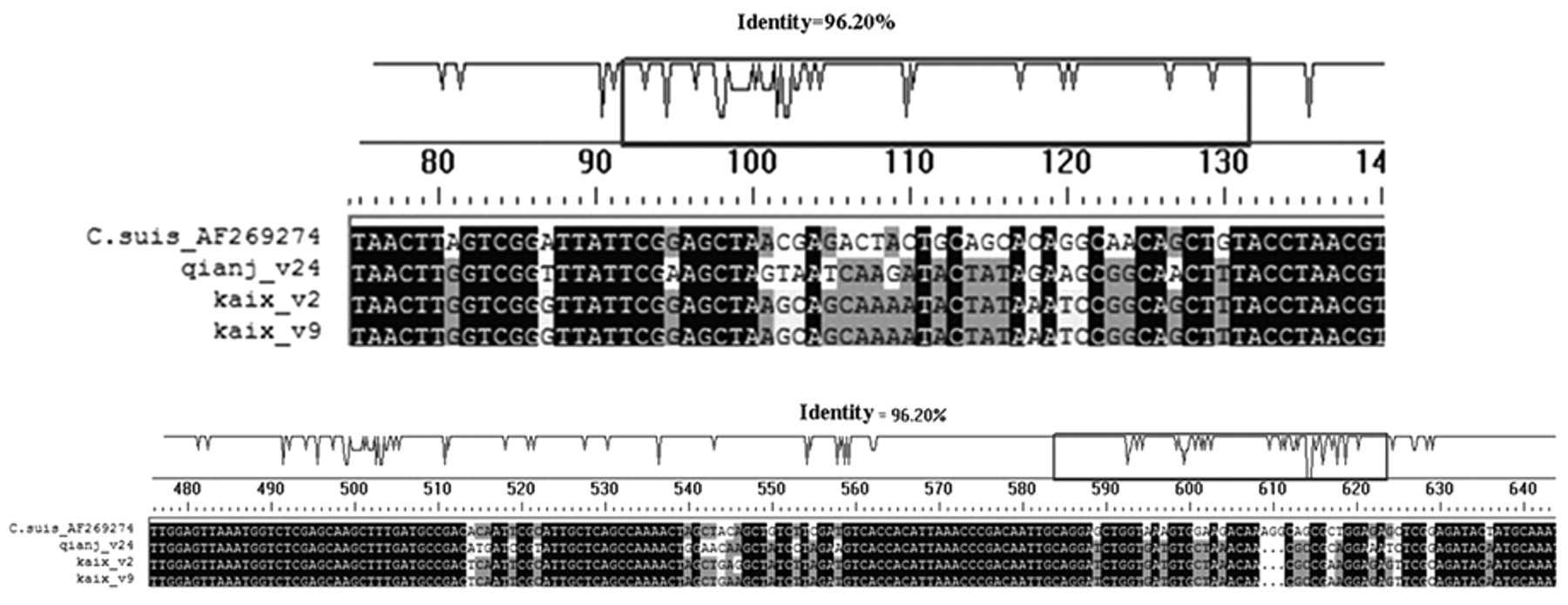
Multi-alignment of the sequences of Chlamydia suis isolates and reference strain (C. suis S45 strain, AF269274). Compared to the S45 strain, the isolates contained triple-base deletion (609–611 bp) and other minor differences.
Discussion
Primer design is the most critical part of PCR-based diagnostic techniques. 8 The nested PCR strategy shows a significant advantage in both sensitivity and specificity. 1,17 The specificity of the nmPCR assay was confirmed by the evaluation of 14 bacterial pathogens in the test of specificity. The nmPCR assay possessed good specificity even in the detection of multiple targets. In the nmPCR, the concentration of magnesium, primers, and dNTP were optimized followed by a step-by-step protocol previously described. 15 To minimize false-negative results, the amplification efficiency of the nmPCR assay was optimized to be individually equivalent for each of Ch. abortus, C. suis, and Ch. pneumoniae. Thus, the nmPCR assay could not miss the target due to the efficiency bias. To evaluate the sensitivity of the nmPCR assay, 2 other PCR assays were used to detect the serially diluted live organism. Owing to the nested amplification of the targets, the sensitivity of the nmPCR assay was greatly elevated.
Swabbing is a harmless and efficient sampling method in chlamydial diagnosis. 12 However, it was noted that inhibitors contained in swabs might generally decrease the activity of DNA polymerase enzyme. 6 Therefore, the direct extraction method (protocol 2) was 10 times more efficient than the silicon column–based method (protocol 1). Such observation might be explained by the relative long time centrifugation, which can gather more chlamydial agents, thus avoiding the loss of DNA as in protocol 1. 9 Compared with the traditional methods, protocol 2 was simpler and easier to perform at a significantly lower cost.
In pathogen isolation, Ch. abortus was positive only in 5 cases out of 122 swabs, which was identical to the results of the nmPCR assay. The 23S real time PCR had a lower increase of positive rates compared with nmPCR. This is likely due to the potential inhibitors in the swabs, while nmPCR could avoid the inhibition because of its 2 nested rounds of PCR reaction. The results of the nmPCR assay corresponded with the chlamydial antibody level and clinical status. It is necessary to further evaluate the potential hazard for workers in the pig farming industry from Ch. pneumoniae emerging in pig breeding farms. 5
The reliability of the nmPCR assay was confirmed by sequencing the products. The reason for this phenomenon may be conservative ompA gene sequence among short PCR products. In summary, the nmPCR assay developed in the current study offered a sensitive and specific screening method for the simultaneous detection of Ch. pneumoniae, Ch. abortus, and C. suis in pigs.
Footnotes
Acknowledgements
The authors thank Dr. John Yang (Lincoln University, Jefferson City, Missouri) and Miss Rasha Khalil for revising of the manuscript, as well as Dr. Jizhang Zhou (Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China) for kindly providing the DNA of Ch. abortus strain. Yingguo Li and Yu Wang contributed equally to the work.
a.
American Type Culture Collection, Manassas, VA.
b.
HyClone laboratories Inc., Victoria, Australia.
c.
Omega Bio-Tek Inc., Norcross, GA.
d.
Promega Corp., Shanghai, China.
e.
Abcam Corp., Cambridge, United Kingdom.
f.
Sangon Biotech Corp., Shanghai, China.
g.
Takara Corp., Da liang, China.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants (CSTC 2009CB1012, CSTC 2007AC1002, CSTC 2009BB1181) from the Committee of Science and Technology of Chongqing.