
Abstract
Classical swine fever (CSF) is a highly contagious disease of pigs. Early detection of the Classical swine fever virus (CSFV) in infected animals and routine surveillance is important for a rapid response and control of an outbreak of CSF. The current study investigated whole blood as a clinical specimen for the detection of CSFV by real-time reverse transcription polymerase chain reaction (real-time RT-PCR) in experimentally infected pigs. The virus was detectable in pre-clinical animals in whole blood and in different fractions of blood, including white blood cells, red blood cells (RBC), and serum. Based on an in-vitro binding assay, CSFV is retained in the RBC fraction. Naturally occurring PCR inhibitors of whole blood were shown to inhibit detection, and several commercial RNA extraction kits failed to remove these inhibitors. The commercial blood RNA extraction protocols were modified to achieve optimized single tube and high-throughput 96-well plate RNA extraction that efficiently removed PCR inhibitors from whole blood and enhanced detection of CSFV in experimentally inoculated pigs. This enabled detection 1–2 days earlier than observed using unmodified RNA extraction protocols. The results show effective use of whole blood as a clinical specimen for diagnosis and surveillance of CSF in pre-clinical animals.
Keywords
Introduction
Classical swine fever (CSF), formerly hog cholera, is a highly contagious disease of pigs and wild boar. Classical swine fever virus (CSFV) belongs to the genus Pestivirus in the family Flaviviridae 18,31 and is closely related to Bovine viral diarrhea virus (BVDV) and Border disease virus (BDV), which primarily infect cattle and sheep, respectively. 3,10,19,33 Pestiviruses consist of a linear 12.5-kb single-stranded RNA genome of positive sense polarity encoding a single 3,898 amino acid long polypeptide that is processed by viral and cellular proteases into 12 structural and nonstructural proteins. 19,24,33 Both ends of the CSFV genome contain untranslated regions that are highly conserved and useful as targets for diagnostic quantitative real-time reverse transcriptase polymerase chain reaction (real-time RT-PCR). 23,27,35
Diagnosis of CSF based on clinical signs can be challenging depending upon the virulence of the CSFV strain and the age and susceptibility of exposed pigs. Recently, highly virulent viruses, responsible for fatal hemorrhagic forms of CSF, have been replaced by CSFV strains of low to moderate virulence, which typically produce subacute, chronic, and/or subclinical illness in pigs, making the clinical diagnosis or field recognition of CSF more difficult. 29 Given the highly contagious nature of CSFV, early detection is critical to limiting the spread of the disease. 16 Since virus concentrations in pre-clinical animals are low, sensitive PCR-based methods are ideal for surveillance and routine diagnosis of CSF in infected herds. 7,12,23,27,35 Other diagnostic methods such as virus isolation and antigen enzyme-linked immunosorbent assay (ELISA) are reliable but time consuming and considerably less sensitive than real-time RT-PCR. 13,17
One of the challenges to PCR-based diagnosis has been reduced efficiency and false-negative results due to the presence of PCR inhibitors in clinical samples. 26,34 Some of the known PCR inhibitors found in clinical specimens include heme, lactoferrin, and immunoglobulin in blood, 1,2 myoglobin and melanin in tissues, 4,14 bile salts and complex polysaccharides in feces, 22,25 and urea in urine. 20
In infected pigs, CSFV can be diagnosed from a wide variety of clinical specimens including blood, tonsil, nasal swab, lymph node, spleen, liver, and kidney. 11,27,32 Blood has been shown to be the one of the most appropriate samples for detection of CSFV by real-time RT-PCR in infected pigs. 21 In addition to being a convenient clinical specimen, blood represents a preferred sample for the diagnosis of CSF due to prolonged viremia during the course of the disease. 13,21,22 In the current study, blood was used as a clinical specimen for detection of CSFV in experimentally infected pigs, and RNA extraction methods were optimized to improve real-time RT-PCR detection.
Materials and methods
Virus strains and animal experiments
Two CSFV strains, the highly virulent CSFV-Brescia and the moderately virulent CSFV-Haiti, were used in the current study. Both viruses were obtained from the U.S. Department of Agriculture, Foreign Animal Disease Diagnostic Laboratory repository. Swine kidney (SK-6) cells were used for growth and propagation of CSF viruses. The SK-6 cells were grown and maintained on Eagle minimal essential media (EMEM) a supplemented with 10% fetal bovine serum (FBS) b at 37°C and 5% CO2. To determine the virus titer, SK-6 cells were grown in 24-well plates, c inoculated with10-fold serial dilutions of the virus in EMEM supplemented with 4% FBS, and incubated at 37°C for 72–96 hr. Viral plaques were detected by an immunoperoxidase assay using the CSFV monoclonal antibody V3 d,15 and a commercial peroxidase system. e Virus titers were expressed as 50% tissue culture infectious dose (TCID50) per ml.
Animal experiments were carried out according to the guidelines and protocols set forth and approved by the Institutional Animal Care and Use Committee. Yorkshire pigs (n = 18) between 8 and 12 years of age and weighing between 18 and 23 kg were inoculated with the virus. Three animal studies were performed, each with 6 pigs, 3 inoculated with CSFV-Haiti and 3 with CSFV-Brescia. Pigs were inoculated either intramuscularly (CSFV-Haiti) or intranasally (CSFV-Brescia) with 200 µl of CSFV-infected ethylenediamine tetra-acetic acid (EDTA)-blood as the inoculum, while control animals received no treatment and were kept isolated from the inoculated animals. The virus titers of the inocula were 104.5 TCID50/ml for CSFV-Haiti and 1010.5/ml for CSFV-Brescia.
Sample collection and processing
Blood was collected from control pigs (uninoculated) or from inoculated pigs on 0, 1, 2, 3, and 5 days post-inoculation (dpi). Whole blood was collected in 5-ml plastic EDTA tubes, f and whole blood for serum was collected in 10-ml plastic blood serum tubes. f Standard protocols were used for fractionation of whole blood into red blood cell (RBC), white blood cell (WBC), or buffy coat, serum, and plasma as outlined in the diagram shown in Figure 1. Two different protocols were used to separate RBC and WBC from blood. In the first protocol, the RBC and WBC suspension (200 µl), derived from whole blood after centrifugation (Fig. 1), was mixed with 5 volumes (1,000 µl) of RBC lysis buffer EL, g incubated on ice for 10 min, and then centrifuged at 1,500 × g for 10 min. The supernatant containing the RBC lysate was collected, and the pellet containing enriched WBCs was resuspended in 2 volumes (400 µl) of RBC lysis buffer to complete the lysis of residual RBCs. The WBC fraction (pellet) collected after the second RBC lysis step was washed twice in PBS (500 µl), and then suspended in 200 µl of PBS for later use. In the second protocol, whole blood (200 µl) and washed RBC and WBC suspension (200 µl) were diluted to 10 ml with PBS and carefully layered over 3 ml of Ficoll h in a 15-ml Falcon tube and centrifuged at 1,000 × g for 30 min at 4°C. The pellet containing enriched RBCs was collected, washed 3 times with 1 ml of PBS, and resuspended in 200 µl of PBS, and used immediately or stored in single use aliquots at –70°C for later use (Fig. 1).
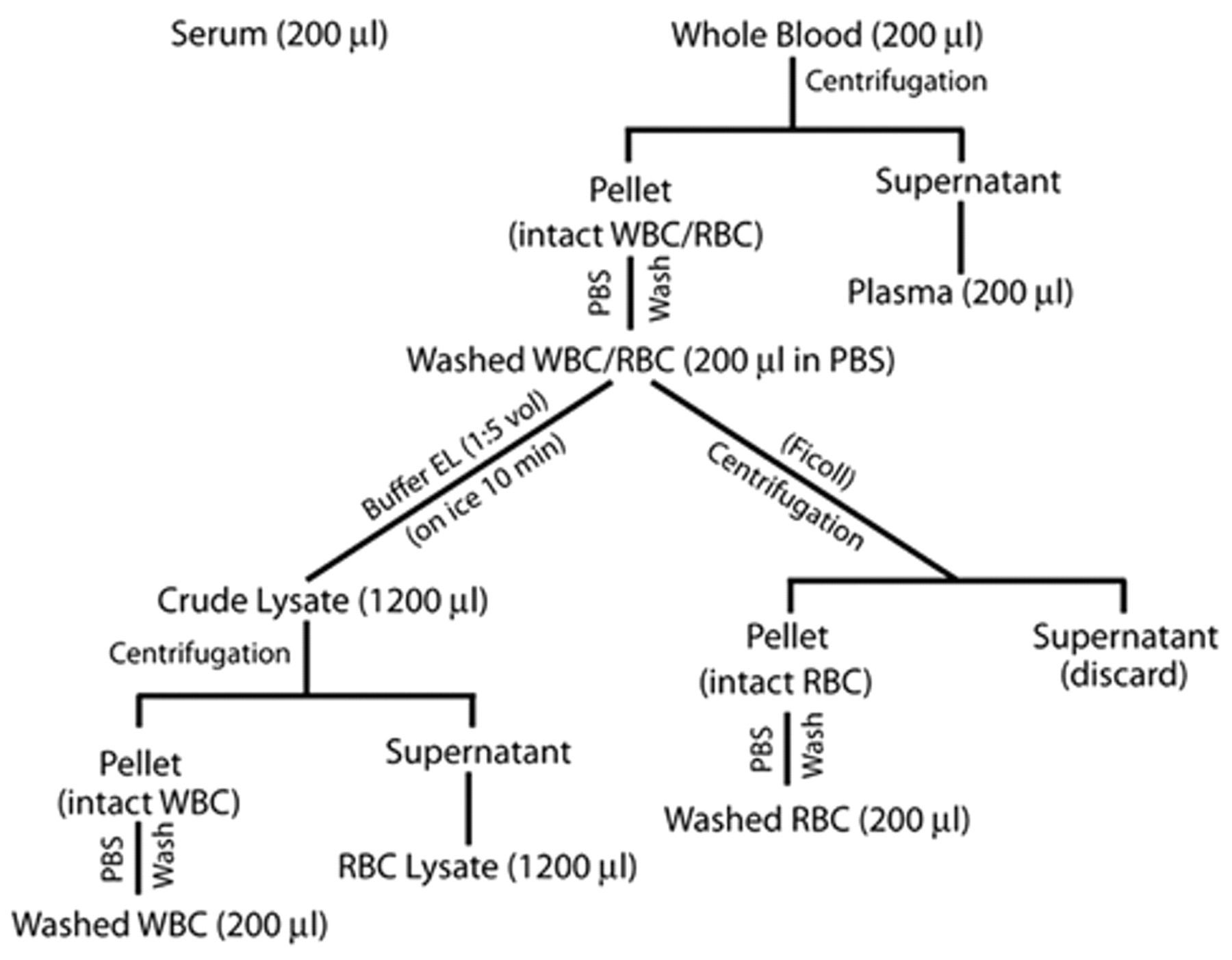
Flow-sheet diagram of the protocol used for fractionation of whole blood into white blood cells (WBC), red blood cells (RBC), serum, and plasma.
Red blood cell Classical swine fever virus binding study
In the current study, 200 µl of whole blood and the purified RBCs derived from whole blood of 2 healthy pigs were inoculated with cell culture–grown CSFV-Haiti or CSFV-Brescia to final concentrations of TCID50 103.8/ml and 108.3/ml, respectively, incubated at 37°C for 1 hr, and fractionated on Ficoll gradient as described above (Fig. 1). Pellets containing RBCs were collected and washed 3 times with 1 ml of PBS and then resuspended in 200 µl of PBS. The above fractionation and washing steps were repeated a minimum of 5 times until the washed RBCs were free of unbound virus particles as assessed by real-time RT-PCR.
Armored RNA
The initial optimization of RNA extraction was carried out using a CSFV-mimic armored RNA, i which is an in-vitro MS2 phage virion packaged synthetic RNA of 489 nucleotides that contains complementary sequences of the forward and reverse primers and the internal probe for the CSFV-specific real-time RT-PCR assay. The above real-time RT-PCR assay amplified a 92–base pair PCR product from either the armored RNA or CSFV genomic RNA. Armored RNA was supplied at a concentration of approximately 1.2 × 1011 copies per µl, which was further diluted to 2.2 × 107 copies per µl in Dulbecco minimum essential media. a
RNA extraction kits and protocols
RNA extractions were performed using whole blood or different fractions of whole blood (WBC, RBC, serum, or plasma) as starting material. For initial evaluation, whole blood from control pigs spiked with serial 10-fold dilutions of armored RNA or live CSFV was used as starting material. Eight commercial RNA extraction kits were used in the study, 6 were single-tube spin column–based kits including ZR Whole-Blood Total RNA kit j (ZR); QIAamp® MinElute Virus Spin kit g (QM); QIAamp® Viral RNA Mini kit g (QV); QIAamp® RNA Blood Mini kit g (QR); Aurum™ Total RNA Mini kit k (AR); and PureLink™ Total RNA Blood kit l (PL); and 2 were magnetic beads–based kits including MagMAX™-96 Whole Blood RNA Isolation kit m (MM) and MagneSil™ Total RNA Isolation kit n (MS). It should be noted that the single-tube RNA kits including AR, QR, and PL failed to use whole blood or intact RBCs but could use WBCs, RBC lysate, and serum as starting material. To optimize the sample volume for efficient RNA extraction, 30-µl, 50-µl, and 100-µl samples were tested for single-tube RNA kits, and 30-µl and 50-µl samples were tested for the magnetic beads RNA kits. Phosphate buffered saline was added to adjust samples to kit-recommended starting volumes prior to extraction. For efficient elution of RNA, the recommended elution buffers were used, and the volumes of the elution buffers tested were 30 µl and 50 µl for AR, QR, ZR, PL, MM, and MS and 30, 50, and 100 µl for QM and QV. All RNA extractions were performed according to the manufacturer’s instructions except for AR and QV. For AR, the kit does not contain RBC lysis buffer, and, therefore, buffer EL g was used to obtain the RBC lysate and WBC fractions (Fig. 1), followed by subsequent use of the manufacturer’s instructions. For QV, the sample volume was adjusted to 200 µl with PBS and then 20 µl of proteinase K g (QV) and 200 µl of the kit-supplied AVL lysis buffer (QV) supplemented with 28 µg of the kit-supplied carrier RNA (QV) were added, and the whole content was mixed and incubated at 56°C for 15 min to complete cellular lysis. Next, 250 µl of 96–100% ethanol was added to the crude lysate, and the entire content applied onto the kit-supplied spin column and centrifuged. In subsequent steps, the spin column with bound RNA was washed twice with 500 µl of high-salt wash buffer AV1, once with 500 µl of low-salt wash buffer AV2, and finally with 500 µl of 96–100% ethanol, and the washed RNA was eluted from the column with 50 µl of the kit supplied elution buffer AVE. An additional washing step between the low- and high-salt washes with a customized high-salt wash buffer consisting of 2 M sodium chloride plus 2 mM EDTA was also employed to remove PCR inhibitors.
Real-time and conventional RT-PCR
The CSF real-time RT-PCR assay used in the current study was developed at the Plum Island Animal Disease Center (PIADC) with primers and probes designed from the highly conserved 5’-untranslated region (UTR) of the CSFV genome (Eberling A, Bieker, J, Reising M, et al., Development, optimization, and validation of a Classical swine fever virus real-time reverse transcription polymerase chain reaction assay. Submitted). The nucleotide sequences of the primers and probe consisted of TGCCCAAGACACACCTTAACC (forward primer); GGCCTCTGCAGCGCCCTAT (reverse primer); and FAM-TGATGGGAGTACGACCTG-MGBNFQ (probe). The real-time RT-PCR reactions were carried out using a one-step commercial kit. g The composition of the reaction master mix (25 µl/per assay) included 0.2 µM forward primer, 0.4 µM reverse primer, 0.2 µM probe, 1 µl of RT-PCR enzyme mix, 12.5 µl of the kit-supplied 2× buffer, 0.5 µl of ROX reference dye 1 (1:10 dilution), 2.5 µl of template RNA, and required amount of nuclease-free water to adjust the volume of the master mix to 25 µl. The thermocycling conditions for amplification include 1 cycle of reverse transcription at 50°C for 30 min, 1 cycle of heat activation at 95°C for 15 min, followed by 45 cycles of PCR amplification with each cycle consisting of a heat denaturation step at 94°C for 15 sec and an extension step at 56°C for 60 sec. Amplifications were performed on a thermocycler o in standard mode, and fluorescence data were collected during the extension step (56°C) in the FAM channel.
Amplifications were also carried out by a conventional RT-PCR assay using a commercial kit. l The RT-PCR master mix had the same composition as that used for the PIADC CSF real-time RT-PCR assay but without the probe and the reference dye (ROX). Thermocycling conditions for RT-PCR included 1 cycle of reverse transcription at 55°C for 30 min, 1 cycle of heat denaturation at 95°C for 2 min, followed by 35 cycles of PCR amplification with each cycle consisting of 5 sec of heat denaturation at 95°C, 30 sec of annealing at 60°C, and 15 sec of extension at 72°C. At the end, a final extension of amplification at 72°C for 2 min was included. All amplification reactions included a positive amplification control (PAC), a positive extraction control, a negative extraction control, and a no template control to ensure the accuracy of the assay. All real-time RT-PCR assays were performed in triplicate, and the results were reported with standard deviations as described in legends to the figures and tables.
Early detection of Classical swine fever virus in blood from experimentally infected pigs
To determine the effectiveness of the modified RNA kits (QV-M and MM-M) on early detection of CSFV in the infected blood, an animal experiment was carried out with 3 pigs inoculated with CSFV-Haiti and 2 pigs inoculated with CSFV-Brescia. The whole blood was collected from the infected animals on 1, 2, and 3 dpi, and the RNA was extracted by the standard and modified QV and MM RNA kits and analyzed by real-time RT-PCR as described above.
Results
Sample selection and characterization of RNA extraction kits
Commercial RNA extraction kits selected for evaluation were divided into 2 types: type I RNA kits including ZR, QV, QM, MM, and MS that recommend whole blood as well as WBC, RBC, RBC lysate, and serum fractions as starting material; and type II RNA kits including QR, AR, and PL that recommend use only with WBC, RBC lysate, or serum fractions but not with whole blood or intact RBC. Using different sample and elution volumes for RNA extraction, optimum results with higher sensitivity of detection of the virus were obtained using 30 or 50 µl of sample volume and 50 µl of elution volume for all RNA kits (results not shown).
Relative distributions of Classical swine fever virus in different fractions of whole blood
Whole blood collected on 5 dpi from pigs infected with CSFV-Haiti and CSFV-Brescia was fractionated into WBC, RBC, serum, and plasma (Fig. 1), and the RNA was extracted from each fraction by MM and analyzed by real-time RT-PCR. Based on the threshold cycle (Ct) values of the RNA extracted from different fractions, higher concentrations of the virus were detected in the whole blood followed by WBC, RBC, and serum (Table 1), suggesting that whole blood is the most appropriate sample type for detection of CSFV in infected animals. Very similar distributions of virus were found in both serum and plasma fractions of infected blood (results not shown); therefore, only results from the serum fractions are presented. One of the interesting findings of the study is the detection of CSFV in the RBC fraction albeit somewhat less sensitive than from whole blood, and WBC fraction. To confirm this, the infected whole blood (5 dpi) was fractionated on Ficoll gradient (Fig. 1), and the purified RBC fraction was extracted and analyzed by real-time RT-PCR. The results show significant and persistent levels of CSFV detected in RBC fractions (Table 2).
Relative distributions of Classical swine fever virus (CSFV) in different fractions of whole blood from experimentally infected pigs.
The threshold cycle (Ct) values of the RNA extracted from each fraction are the average of 3 replicates with standard deviations as shown.
Detection of Classical swine fever virus (CSFV) in the red blood cell fractions of infected whole blood after separation by density gradient centrifugation on Ficoll gradient.*
QV = QIAamp® Viral RNA Mini kit g ; MM = MagMAX™-96 Whole Blood RNA Isolation kit. m *WBC = white blood cells; RBC = red blood cells;
The threshold cycle (Ct) values shown in the table are the average of 3 replicates with standard deviations as shown.
To explore the possibility of nonspecific association between free live virus and RBCs, whole blood from healthy pigs and the Ficoll gradient–purified RBCs were inoculated with cell culture–grown CSFV-Haiti and CSFV-Brescia, and the content repeatedly fractionated on Ficoll gradients. Analysis by real-time RT-PCR (Table 3) on the RNA extracted from RBCs at various levels of purification show presence of the virus (CSFV-Haiti or -Brescia) in purified RBCs, indicating a tight association of the virus with the RBCs.
In-vitro binding of Classical swine fever virus (CSFV) to purified red blood cells from control pigs.*
RBC = red blood cells; WB = whole blood; TCID50 = 50 percent tissue culture infectious dose; ND = not determined.
The threshold cycle (Ct) values shown in the table are the average of 3 replicates with standard deviations as shown.
Performance evaluation of commercial RNA extraction kits
For proper evaluation of the RNA kits, the performances of the type II RNA kits (QR, PL, and AR) were compared against the performance of the type I RNA extraction kit (MM) using CSFV-Haiti–infected whole blood and different fractions (WBC, RBC, and serum). Analysis of the extracted RNA by real-time RT-PCR indicated higher sensitivity of detection of the virus with the RNA extracted by MM compared to the RNA extracted by QR, AR, and PL (Fig. 2).
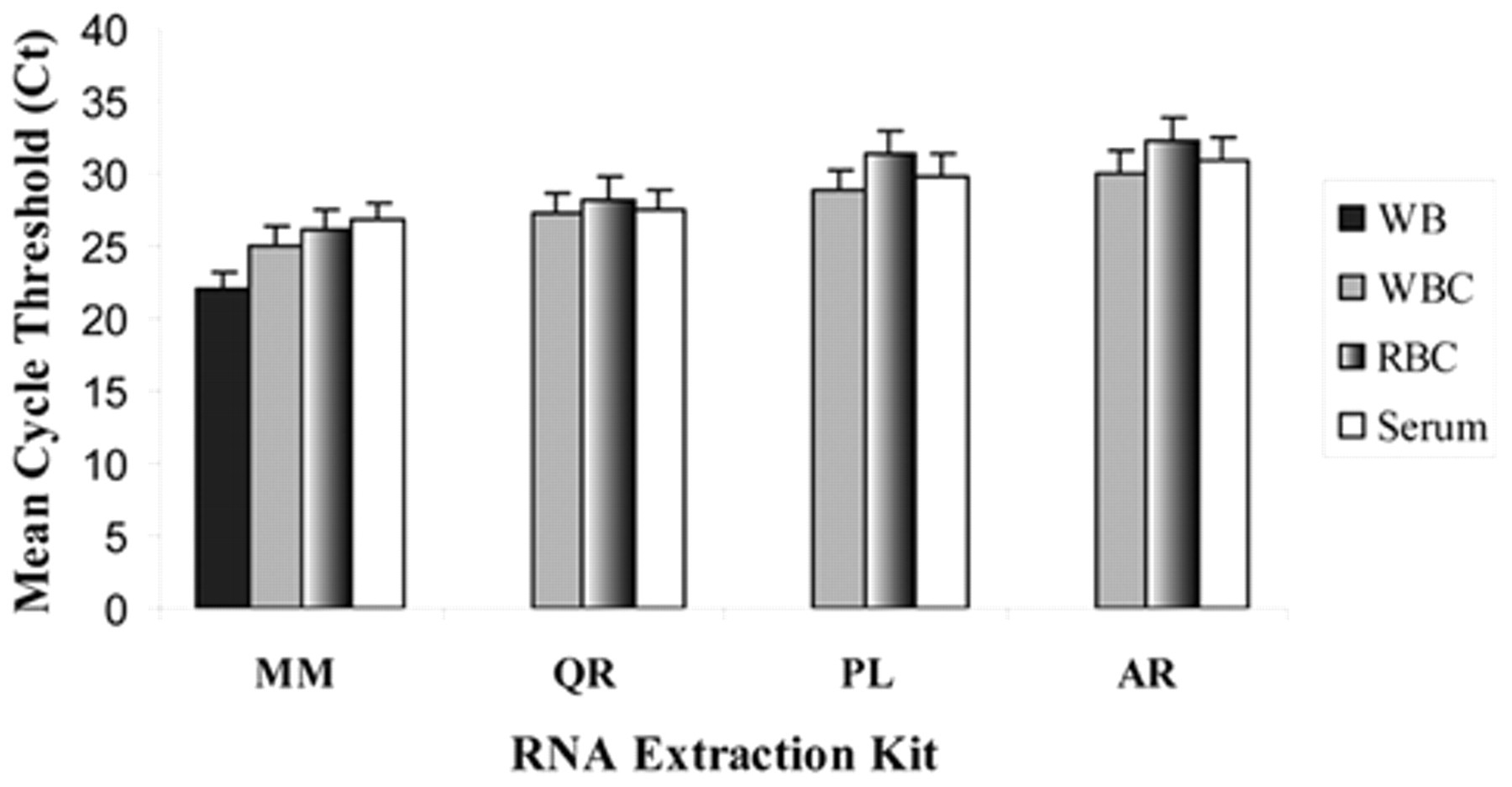
Analyses of the Classical swine fever virus (CSFV) RNA extracted from whole blood and its fractions by type I and type II RNA extraction kits. Whole blood was collected on 5 days post-infection (dpi) from an experimental pig infected with classical swine fever (CSF)-Haiti and fractionated into WBC, RBC (lysate), and serum as described in the materials and methods section. A 30-µl sample from each fraction was extracted by MagMAX™ (MM; type I RNA kit), and QIAamp® RNA Blood Mini kit g (QR), PureLink™ Total RNA Blood kit l (PL), and Aurum™ Total RNA Mini kit k (AR; type II RNA kits), and the extracted RNA were analyzed by real-time reverse transcription polymerase chain reaction as described in the materials and methods section. The threshold cycle (Ct) values shown in the figure are the average of 3 replicates with standard deviations indicated by error bars.
Since whole blood was found to be the most appropriate sample for detection of CSFV (Fig. 2), attention was focused on subsequent evaluation of the type I RNA kits using whole blood as the starting material. Whole blood from healthy pigs spiked with 10-fold serial dilutions of the armored RNA (working stock: 2.2 × 107 copies/ml) was used as the starting material for RNA extraction. Based on the real-time RT-PCR analysis of the RNA extracted from 30 µl of sample from each dilution, higher sensitivity of detection of armored RNA (lower Ct values) was found with the RNA extracted by MM, ZR, and QV (up to 10−8 dilution) followed by QM (up to 10−6 dilution), and MS (up to 10−2dilution; results not shown).
To determine if there was any real-time RT-PCR inhibition, armored RNA was serially diluted in PBS (control) and whole blood from healthy pigs and used as the starting material for RNA extraction. Three RNA kits (MM, QV, and ZR) were used in the study as they performed better than the other kits (see above). The results of real-time RT-PCR (Table 4) show higher Ct values of the armored RNA extracted from serial dilutions in whole blood than from serial dilutions in PBS, indicating the presence of PCR inhibitors in whole blood, as reported by others. 1,2,11 A similar inhibition profile was observed with the RNA extracted from serial dilutions of CSFV-Haiti in whole blood as compared to PBS as a diluent (results not shown).
Inhibition of polymerase chain reaction by naturally occurring inhibitors of whole blood.*
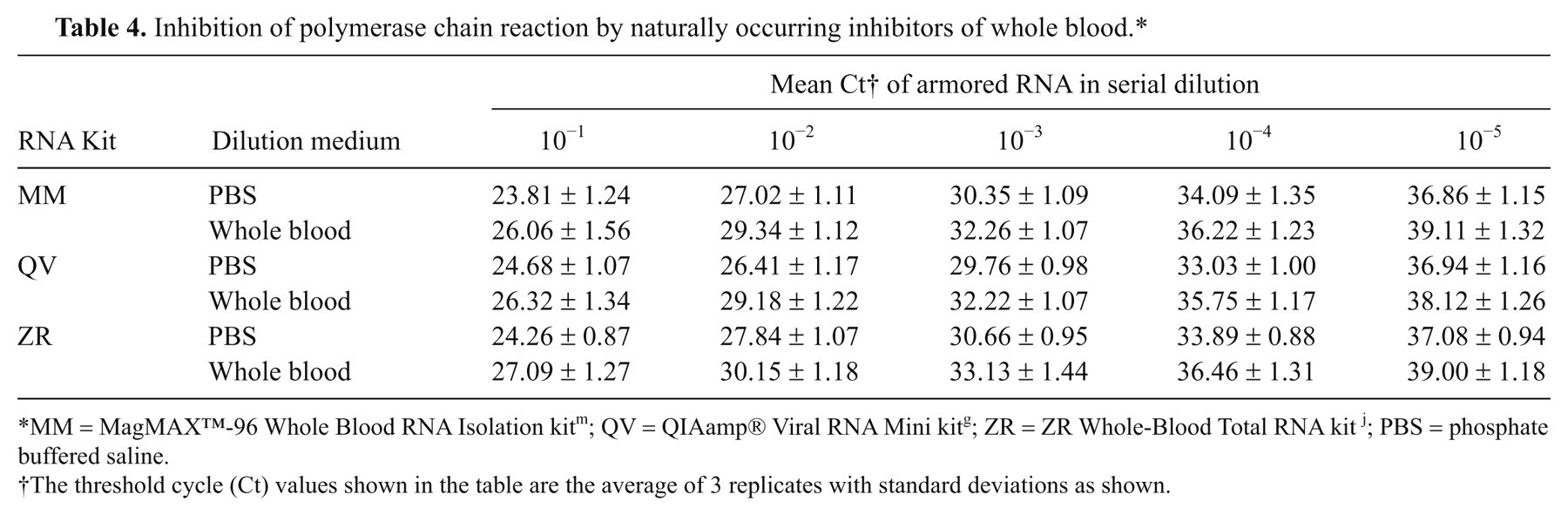
MM = MagMAX™-96 Whole Blood RNA Isolation kit m ; QV = QIAamp® Viral RNA Mini kit g ; ZR = ZR Whole-Blood Total RNA kit j ; PBS = phosphate buffered saline.
The threshold cycle (Ct) values shown in the table are the average of 3 replicates with standard deviations as shown.
Removal of PCR inhibitors from whole blood
To improve the capacity of the commercial RNA kits to remove PCR inhibitors from blood, 3 type I RNA kits (QV, ZR, and MM) were subjected to several modifications as described in the materials and methods. Whole blood from healthy pigs spiked with armored RNA was used as the starting material. Optimum results with higher sensitivity of detection of the armored RNA were obtained using 30 µl of sample volume for QV and ZR, 30 µl or 50 µl of sample volume for MM (results not shown), and 50 µl of elution volume for all RNA kits. Dilution of the RNA extracted from whole blood with RNase-free water did not improve the sensitivity of detection of the armored RNA by real-time RT-PCR (results not shown). Additional washes with kit-supplied wash buffers, including both high- and low-salt wash buffers, also had little effect on removal of PCR inhibitors. Previously, it was shown that a high salt wash buffer consisting of 2 M sodium chloride and 2 mM EDTA significantly improved the sensitivity of detection of avian influenza RNA extracted from fecal samples by a commercial kit. p,8,9 Additional washes with this custom high-salt wash buffer did indeed improved the sensitivity of detection of armored RNA from whole blood extracted with MM, QV, and ZR (Table 5). The effect of DNase I treatment on RNA extraction was also tested. The latter step is part of the standard MM protocol, but it had little effect on the extraction of armored RNA from blood with MM, QV or ZR (results not shown).
Effect of additional high salt washes with 2 M sodium chloride–2 mM ethylenediamine tetra-acetic acid on extraction of armored RNA serially diluted in whole blood.*
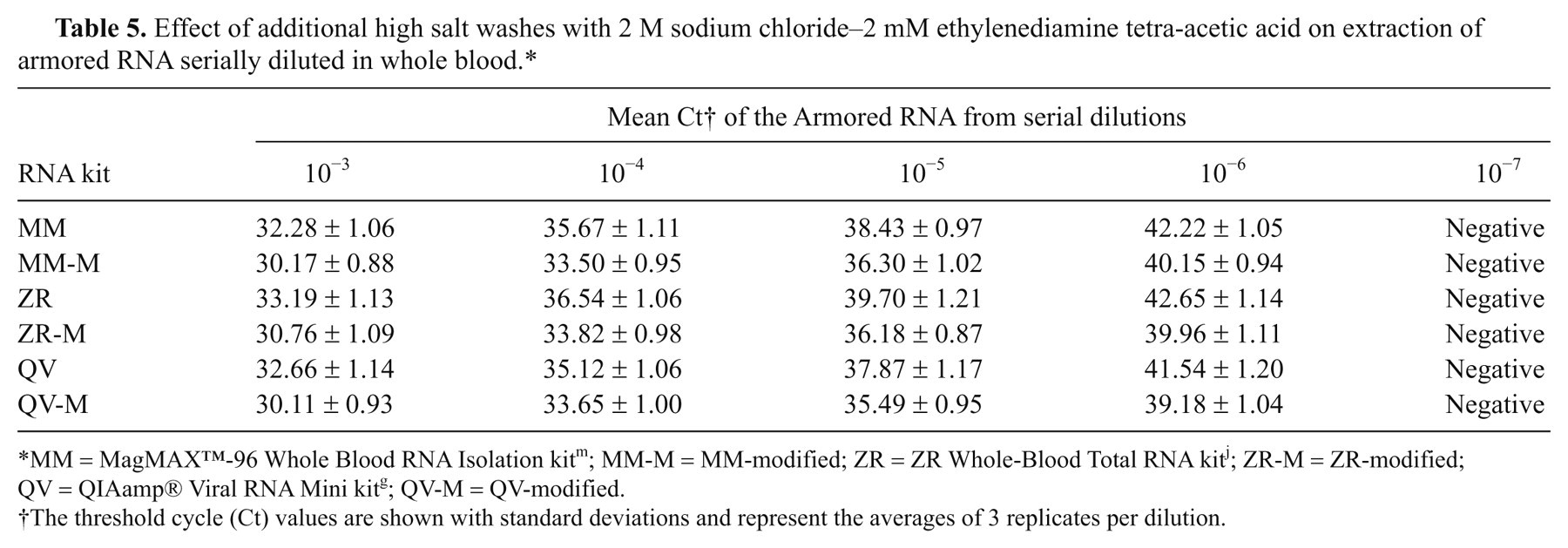
MM = MagMAX™-96 Whole Blood RNA Isolation kit m ; MM-M = MM-modified; ZR = ZR Whole-Blood Total RNA kit j ; ZR-M = ZR-modified; QV = QIAamp® Viral RNA Mini kit g ; QV-M = QV-modified.
The threshold cycle (Ct) values are shown with standard deviations and represent the averages of 3 replicates per dilution.
Finally, the MM and QV RNA kits were tested with and without modification by a supplemental washing step (2 M sodium chloride–2 mM EDTA). These modified RNA extraction kit protocols are designated QV-M and MM-M to distinguish them from the standard commercial kit protocols. CSFV-infected whole blood serially diluted in blood from healthy pigs was employed as starting material for this comparison. The results (Fig. 3) show higher sensitivity of detection of CSFV with RNA extracted by the modified RNA extraction protocols than by unmodified protocols.
Performance evaluation of standard MagMAX™ (MM) and QIAamp® Viral RNA (QV) kits as well as the same kits modified (MM-M and QV-M) by addition of a customized high-salt wash step for extraction of Classical swine fever virus RNA from infected whole blood. Serial dilutions of infected blood were made in whole blood from healthy pigs. A 30-µl sample from each dilution was used as starting material. The extracted RNA was analyzed by real-time reverse transcription polymerase chain reaction (real-time RT-PCR) as described in the materials and methods section. The threshold cycle (Ct) values shown in the figure are the average of 3 replicates with standard deviations indicated by error bars.
Early detection of Classical swine fever virus
Using standard RNA extraction protocols (QV and MM), early detection of virus was found to be 2 dpi and 3 dpi in pigs infected with CSFV-Haiti and CSFV-Brescia, respectively (Table 6). In contrast, early detection of the virus was 2 dpi for both CSFV-Haiti and CSFV-Brescia using modified protocols (QV-M and MM-M) for RNA extraction (Table 6). The clinical signs from CSFV infections including high fever (40–41.6°C), depression, inappetence, diarrhea, and conjunctivitis began to appear after 3 dpi in all experimentally infected animals. Early detection of the virus at 1 dpi in blood occurred at C t values above 40, which are inconclusive according to the standard operating protocol of the PIADC real-time RT-PCR assay. Therefore, PCR products amplified by the inconclusive real-time RT-PCR assays as well as repeat conventional RT-PCR assays were confirmed by gel electrophoresis (data not shown). Hence, the combined results of real-time and conventional RT-PCR confirmed the earliest detection of virus at 1 dpi for pigs infected with CSFV-Haiti and 2 dpi for pigs infected with CSFV-Brescia (Table 6).
Early detection of Classical swine fever virus (CSFV) by real-time reverse transcription polymerase chain reaction in whole blood of experimentally infected pigs with the RNA extracted by standard and modified commercial kits.*
DPI = days post infection; MM = MagMAX™-96 Whole Blood RNA Isolation kit m ; MM-M = MM-modified; QV = QIAamp® Viral RNA Mini kit g ; QV-M = QV-modified.
The threshold cycle (Ct) values shown in the table are average of 2 replicates with standard deviations in the range between 0 and 3%.
Discussion
The results of the current study suggest that whole blood rather than any fraction of whole blood represents the most appropriate sample for early detection of CSFV. In addition to CSFV’s known tropism for lymphoid tissues, the virus has been shown to be predominantly associated with apoptosis of peripheral blood mononuclear cells (PBMC), including the WBCs. 28,29 This can result in a drop in WBC counts by as much as 65.87% at 1 or 2 dpi, in pigs inoculated with CSFV. 36 These findings correlate well with the detection of virus at 1 and 2 dpi in whole blood from experimentally infected pigs reported in this study. In the current study, a significant amount of the virus was also detectable in the RBC fractions. Unlike the WBCs, mature RBCs are non-nucleated and lack the ability to support virus replication, and hence, the detection of CSFV in RBCs is most unexpected. Ficoll gradient studies presented here suggest that this is most likely due to nonspecific but tight binding interactions between the virus and the RBCs, a phenomenon that merits further investigation.
One of the major challenges of PCR-based diagnosis has been the presence of PCR inhibitors in commonly used clinical specimens including blood, urine, tissues, and fecal materials. 26,34 These inhibitors are often co-purified with viral nucleic acids during extractions. 1,2,26 Eight commercial RNA extraction kits were evaluated on whole blood and fractionated blood revealing whole blood to be the best sample choice for CSFV detection by real-time RT-PCR. None of the RNA kits however, were able to completely remove PCR inhibitors from blood. The authors that found the use of smaller sample volumes (30 µl) and an additional custom wash step containing 2 M sodium chloride and 2 mM EDTA significantly improved the capacity of the commercial RNA kits to remove PCR inhibitors from whole blood. RNA extracted from sample volumes greater than or less than 30 µl resulted in either higher C t values or inconsistent results and are therefore not recommended for extraction. The new high salt wash buffer used in the current study was previously shown to effectively remove PCR inhibitors from fecal samples (cloacal swabs) of wild birds, and it has improved the limit of detection of avian influenza virus by real-time RT-PCR using the RNA extracted by MagMAX™-96 AI/ND viral RNA isolation kit from Applied Biosystems. n,8 Herein, we show that the custom high salt wash is equally effective for both single tube and magnetic bead RNA kits from different vendors. Similar high salt, EDTA containing wash buffers have been previously shown to improve detection of viral nucleic acids. 5,14 The EDTA effect may be linked to its ability to chelate metal ions such as iron that have been found to be inhibitory for PCR. 30
The modified RNA extraction protocols used in this study to detect CSFV at 1–2 dpi could help in the early detection of CSFV in pre-clinical animals. For instance, previously reported detection of CSFV in experimentally infected pigs was 2–3 dpi. 6,17,32 These modified protocols should prove useful when blood is under investigation as a clinical sample for detection of CSFV and may be anticipated to improve RT-PCR–mediated detection of other RNA targets from blood.
Footnotes
Acknowledgements
The authors sincerely thank Jeff Babcock and his team from the PIADC for assistance in animal care and inoculation experiments. The authors also thank members of the Proficiency and Validation Services Section of FADDL for helpful discussions and support and Elizabeth Clark of the Foreign Animal Disease Diagnostician School for providing access to animal samples.
a.
Gibco, Grand Island, NY.
b.
Atlas Biologicals, Fort Collins, CO.
c.
Costar, Cambridge, MA.
d.
Cedi Diagnostics, Lelystad, The Netherlands.
e.
Vectastain ABC kit, Vector Laboratories, Burlingame, CA.
f.
BD, Franklin Lakes, NJ
g.
QuantiTect® Multiplex RT-PCR NR kit, Qiagen, Valencia, CA.
h.
Ficoll-Paque™, Sigma-Aldrich, St Louis, MO
i.
Asuragen Inc., Austin, TX
j.
ZYMO Research Corp., Irvine, CA.
k.
Bio-Rad Laboratories, Hercules, CA.
l.
SuperScript IIITM RT-PCR, Invitrogen Corp., Carlsbad, CA.
m.
Applied Biosystems/Ambion, Austin, TX.
n.
Promega Corp., Madison, WI.
o.
ABI 7500, Life Technologies Corp., Carlsbad, CA.
p.
MagMAXTM-96 AI/ND Viral RNA Isolation kit, Applied Biosystems/Ambion, Austin, TX.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the USDA, Animal Plant Health Inspection Service, Veterinary Services, National Veterinary Services Laboratories.