
Abstract
The hemagglutinin-neuraminidase (HN) glycoprotein of Newcastle disease virus (NDV) constitutes, together with the fusion glycoprotein, the main surface antigen of this avian pathogen, which causes a highly contagious disease, relevant economically worldwide. The purpose of this work was to obtain the HN glycoprotein as a soluble antigen in culture supernatants of recombinant baculovirus-infected Spodoptera frugiperda (Sf9) cells and to evaluate its application to the development of a recombinant enzyme-linked immunosorbent assay (rELISA) for the analysis of chicken sera. A transfer vector for baculovirus containing the sequence of a melittin signal peptide was constructed and the sequence coding for HN protein without its own signal peptide was cloned. The recombinant protein was secreted and recovered easily from the culture medium of Sf9-infected cells. The recombinant protein was evaluated as antigen for ELISA coating the plates with the recovered HN using 79 positive and 142 negative samples. The Cohen kappa value resulted 0.91, indicating excellent agreement between the rELISA and the hemagglutinin inhibition tests. The rELISA was also compared with a commercial ELISA, finding high levels of agreement between both assays. The present results show that the cloning strategy developed yielded the HN protein free in the cell culture supernatant and that the recombinant protein retained its reactivity with anti-NDV HN antibodies in chicken sera.
Newcastle disease virus (NDV; family Paramyxoviridae, subfamily Paramyxovirinae, genus Avulavirus) is an avian paramyxovirus that causes an economically important and highly contagious disease of poultry. 1 Newcastle disease virus is an enveloped virus with a nonsegmented negative-stranded RNA genome. The viral particle is constituted of a lipid bilayer membrane bearing hemagglutinin-neuraminidase (HN) and fusion (F) glycoprotein spikes. 1 Both glycoproteins are considered to be involved in the development of specific immunity to infection. 3,20 The F protein is also involved in membrane fusion and virus penetration, while the HN protein allows virus attachment to the host cell receptor.
The hemagglutination inhibition (HI) test is the recommended assay for the detection of anti-NDV antibodies. 2 However, the HI test is a labor-intensive method, and reading of its results is relatively subjective. A previous study using a proficiency test reported that the HI test yielded a higher level of variation than commercial enzyme-linked immunosorbent assay (ELISA) among different laboratories. 8 Nevertheless, the HI test is often referred to and used as the “gold standard” in most studies. Several ELISAs are commercially available for the detection of specific anti-NDV antibodies and their results have been shown to correlate with those obtained by the HI test. Also, several “in-house” ELISAs for the detection of anti-NDV sera have been developed, using whole viral particle antigen 22 or recombinant antigens produced through bacterial 18 or baculoviral expression systems. 9,16 The objective of the present study was to express the HN protein of NDV in a secreted form for use in developing an ELISA for detection of anti-NDV antibodies in chicken sera.
The HN coding region of NDV LaSota strain a was amplified from complementary DNA synthesized from genomic RNA isolated by the TRIzol method, b according to the manufacturer’s instructions. Complementary DNA was obtained using random hexamers, and amplification of HN coding region was carried out using HNf (5’-AAAGGATCCGCTAGCACACCTAGCGAT-3’) and HNr (5’-AGGCTCTAGATTGCCAGACCTGGC-3’) oligonucleotides. In this way, a product of 1,585 bp was amplified (from position 6562 to 8146), excluding the first 150 bp coding for 50 amino acids of the protein predicted as the signal peptide and the transmembrane domain, based on previously reported LaSota nucleotide sequence. 7 The forward primer contained a BamHI restriction site, and the reverse primer contained an XbaI restriction site. Utilizing these restriction sites, the amplified fragment was cloned into plasmid pBacAV5. The pBacAV5 consisted in a pBacPAK8 backbone c where the honeybee melittin signal sequence (which can enable efficient translocation of proteins into the endoplasmic reticulum of Spodoptera frugiperda cells 21 ) was cloned upstream of the multiple cloning site to substitute the viral signal peptide; the V5 epitope tag sequence (excised from the pIB/V5-His plasmid d ) was cloned downstream of the multiple cloning site, to obtain a fusion protein with the cloned sequence of interest, easily detectable by Western blot assays using an anti-V5 monoclonal antibody e (Fig. 1A). The polymerase chain reaction (PCR) product was cloned into the transfer plasmid, and recombinant plasmids were isolated following classical molecular biology protocols. 17 The Autographa californica multiple nucleopolyhedrovirus f (AcMNPV) was used as a vector for the expression of HN protein. Spodoptera frugiperda (Sf9) cells were co-transfected with the recombinant plasmid and linearized the AcMNPV DNA, using a commercial transfection kit f following the manufacturer’s instructions. Recombinant baculoviruses were purified by 2 rounds of final end-point dilution, 19 and selected clones were amplified by 3 passages in Sf9 insect cells. Finally, the viral stock (AcMNPV/HN) was titrated, and HN expression was characterized by Western blot assay. For virus production, monolayers of Sf9 cells were infected with the viral inoculums at a multiplicity of infection (moi) of 5. All procedures were performed as previously described. 19 After 72 hr, cells were collected, washed with phosphate buffered saline (PBS; pH 6.2) and resuspended in cracking buffer. 12 The supernatants of infected cells were also retained to evaluate HN expression in culture medium. Samples of cellular extracts and supernatants were analyzed by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and the presence of the recombinant protein was confirmed by Western blot assay 12 using a rabbit anti-HN polyclonal serum. g Figure 1B shows the presence of a double band slightly lower than the band observed with purified NDV. Both cell extracts and supernatants of infected Sf9 cells showed the same band patterns. The double-band pattern was previously reported for HN expressed in insect cells by the baculovirus system, 4 and it corresponds to the result of glycosylation heterogeneity, as it has been reported for several glycoproteins synthesized in the baculovirus system. 19 These results indicated the proper expression of the recombinant HN protein by the baculovirus expression system. Also, the presence of the protein in the supernatant demonstrated that the cells secreted the protein into the culture medium.
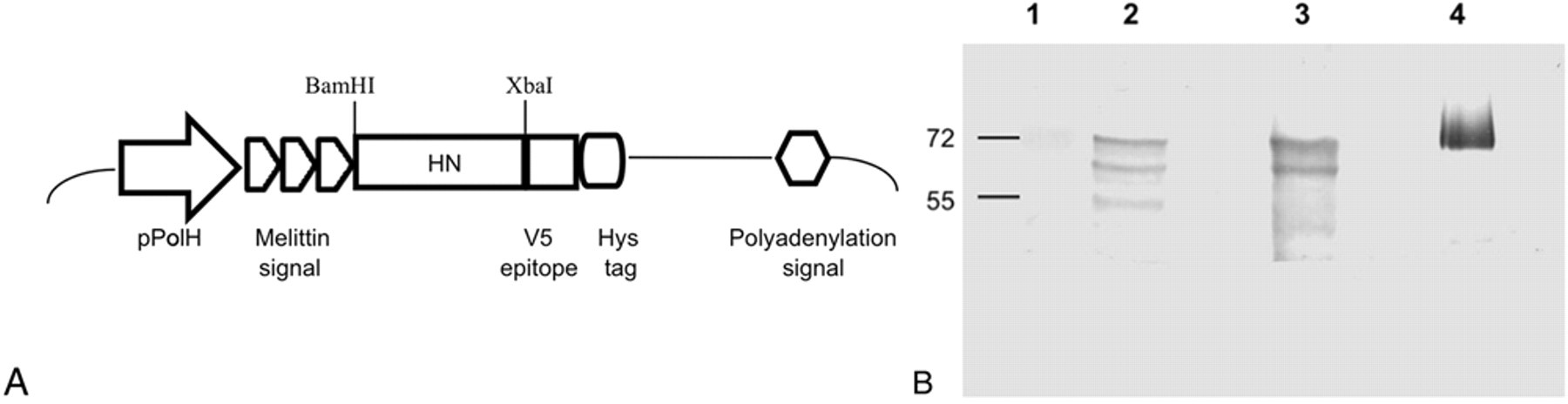
Production of the recombinant hemagglutinin-neuraminidase (HN) protein.
For the production of the recombinant antigen to be used in the recombinant ELISA (rELISA), monolayers of Sf9 cells grown in Sf900 medium were infected with the viral inoculum at a moi of 5 in supplemented Sf900 medium (10% fetal bovine serum, d 1% antibiotic–antimycotic d ). After 24 hr at 27°C, the medium was replaced by fresh medium without serum and incubated for 72 hr at 27°C. Then, supernatant fluid was collected, centrifuged for 15 min at 290 × g and concentrated 4 times utilizing centrifugal filter devices. h An aliquot was resuspended in cracking buffer, and the presence of the recombinant protein was evaluated by Western blot assay as previously described. 12
Optimal dilutions of the recombinant antigen as well as chicken sera and goat anti-chicken immunoglobulin G antibodies conjugated with horseradish peroxidase i were determined by checkerboard titration. Positive and negative sera were employed in order to establish the optimal conditions of each step of the assay. Twenty specific pathogen–free (SPF) chickens j were immunized with anti-NDV vaccine, k and sera were obtained at 28 days postvaccination. Twenty unvaccinated SPF chickens were bled to obtain the negative sera. Positive and negative samples were pooled, tested by HI as previously described 14 and by conventional ELISAs, 5 and then were used for setting the optimal assay conditions. In order to verify the absence of nonspecific reactions, the positive and negative serum pools were tested in the absence of HN antigen (supernatant of mock-infected Sf9 cells). Briefly, 3-fold serial dilutions of the recombinant antigen (and the negative control antigen) in carbonate buffer saline were tested. As shown in Figure 2, 1:27 (which corresponded to 40 ng/well as estimated by comparison with a standard curve of bovine serum albumin stained with Coomassie brilliant blue) was determined to be the optimal dilution of the recombinant antigen in the rELISA. Furthermore, the assay revealed the absence of nonspecific reactions with both the positive and negative sera. When used at 1:27 optimal dilution, recombinant antigen from one 150-cm2 flask of Sf9-infected cells was sufficient to coat 33 96-well ELISA plates. Dilutions of serum and conjugate, as well as composition of blocking solutions, were also evaluated by a similar checkerboard titration protocol (data not shown).
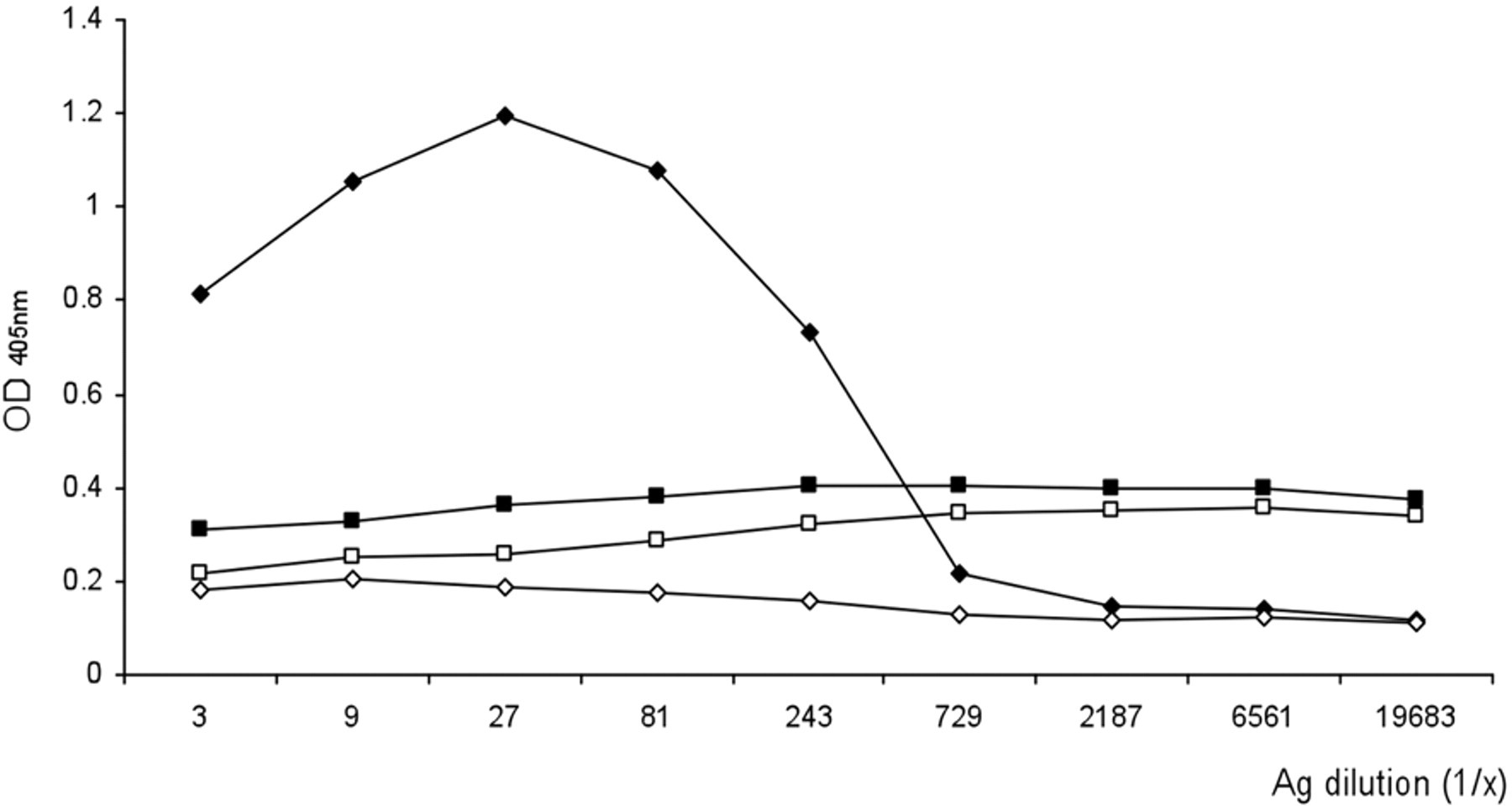
Determination of the optimal dilution of the recombinant antigen by enzyme-linked immunosorbent assay. Three serial fold dilutions of the antigen (supernatant of Autographa californica multiple nucleopolyhedrovirus/hemagglutinin-neuraminidase protein Spodoptera frugiperda [Sf9]-infected cells or of mock-infected Sf9 cells) were evaluated against positive and negative control sera diluted 1/50. Black square indicates negative antigen/positive serum; white square indicates negative antigen/negative serum; black diamond indicates positive antigen/positive serum; and white diamond indicates positive antigen/negative serum. OD = optical density.
The final rELISA protocol utilized microtitration plates l coated with 40 ng/well of the recombinant HN protein in carbonate buffer (pH 9.6), by overnight incubation at 4°C. After each step, plates were washed 5 times with washing buffer (PBS and 0.05% Tween-20). After removing the excess of the unbound antigen by washing, a blocking step was performed for 1 hr at 37°C with PBS, 0.05% Tween-20, 5% horse serum (PBS-T-HS), 5% non-fat dried milk. Then, a 50-µl volume of an optimal dilution (1:50) of chicken sera in PBS-T-HS was added to the plate and incubated for 1 hr at 37°C. Finally, 50 µl of a 1:2,000 dilution (25 ng/well) of horseradish peroxidase–conjugated antibody i in PBS-T-HS were applied for 1 hr at 37°C. A 50-µl volume of the substrate solution (2,2’-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) 0.5 mg/ml m in 0.1 M citrate buffer [pH 5] containing 0.03% hydrogen peroxide) was added, and optical density (OD) was read at 405 nm after an incubation of 20 min at room temperature. Each sample was tested in duplicate and the mean value was determined.
Sets of sera from vaccinated (with inactivated or live attenuated vaccines based on LaSota strain of NDV) and nonvaccinated SPF White Leghorn chickens were used to evaluate the rELISA. Seventy-nine sera from vaccinated birds, obtained at different times post-immunization (14, 21, 28, 35, and 45 days post vaccination), and a total of 142 negative sera were evaluated. The positive threshold value (0.316 OD units), calculated as the mean OD value of the negative sera plus 3 standard deviations (Xneg + 3σ), was used to estimate the sensitivity (Se) and specificity (Sp) of the rELISA assay 13 :

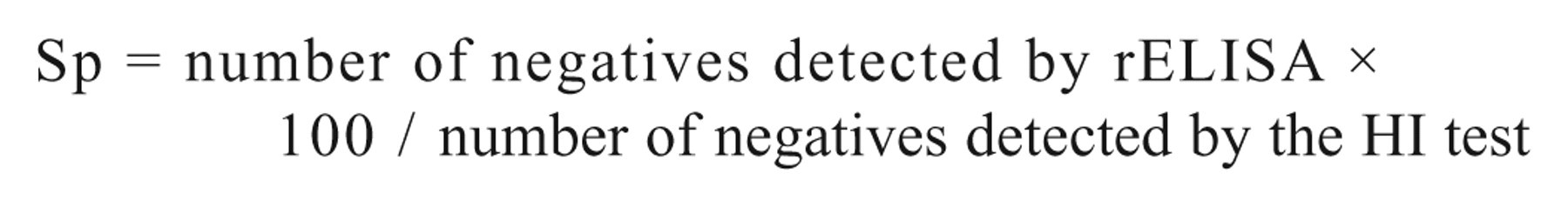
Intra- and interassay variation coefficients (VCs) were estimated as previously described, 13 where

The Se and Sp of the rELISA were evaluated in comparison to the HI test as the standard method for NDV antibody detection, using a total of 221 serum samples. Since 7 out of the 79 positive samples tested were negative by rELISA (Table 1), the estimated Se of the rELISA was 91.14%. On the other hand, 2 out of the 142 negative samples evaluated were positive by rELISA, achieving a Sp value of 98.6%. In order to measure the percentage of agreement between both assays, the Cohen kappa statistic was calculated. The κ value [(Po − Pe)/(1 − Pe)], where Po = (72 + 140)/221 and Pe = (74 × 79 + 147 × 142)/2212, resulted in 0.91, indicating excellent agreement between the rELISA and the HI test. 10 The repeatability results of the rELISA were 5.6% and 7.7% for intra- and interassay VCs, respectively, indicating adequate repeatability at this stage of the development of the assay.
Comparison of recombinant enzyme-linked immunosorbent assay (rELISA) and hemagglutination inhibition (HI) test for detection of Newcastle disease virus (NDV) antibodies in chicken sera.*

Sensitivity: 91.14%; specificity: 98.6; relative accuracy: (72 + 140/221) × 100 = 95.9%; κ value: 0.91
When 199 out of the 221 serum samples were evaluated by the rELISA in comparison with a commercial ELISA for the detection of anti-NDV antibodies, high levels of agreement between both assays were observed. Out of 124 negative sera (determined by the SVANOVIR test n ), 119 were also negative by the rELISA. The other 5 samples (negative by SVANOVIR and positive by the rELISA) yielded OD values just above the 0.316 cut-off value (mean OD of the 5 samples = 0.373). On the other hand, when 75 positive samples (by SVANOVIR) where analyzed by the rELISA, 64 were also positive by rELISA, but 11 were negative. Since the commercial ELISA detects antibodies against whole NDV and the discrepant samples were collected on day 14 post-vaccination when seroconversion is just starting, it is possible to speculate that early in the antibody response, antibodies directed against proteins other than HN might be present in those samples.
Two groups have previously reported the development of ELISAs based on recombinant NP protein obtained directly from insect cells, using soluble fractions obtained after freeze thawing and centrifugation 9 or soluble fractions from sonicated cell lysates 16 as the antigen. Although those investigators reported no interference from background cellular proteins, when working with cells, a purification or at least a semipurification step is usually needed to ensure optimal performance. 6,11,15 In the present report, the HN protein of NDV was expressed by the baculovirus expression system and it was used directly for the development of a rELISA. The protein obtained was designed to be secreted into the cell culture supernatant and therefore needed no purification steps, thus rendering the technique a more economic and convenient tool. The relatively high agreement between the rELISA developed in the present study and the HI test (kappa = 0.91) indicates that the new assay might be an appropriate alternative for the detection of NDV-specific antibodies in chicken sera. The economic benefit that could be obtained using the recombinant protein, may allow the rELISA to be used in developing countries for NDV surveillance and diagnostic.
Footnotes
a.
National Service for Animal Health, Argentina (SENASA).
b.
TRIzol® Reagent, Life Technologies, Grand Island, NY.
c.
Clontech, Mountain View, CA.
d.
Invitrogen, Carlsbad, CA.
e.
Amersham, Buckinghamshire, United Kingdom.
f.
Pharmingen, San Diego, CA.
g.
Rabbit anti-HN polyclonal serum, kindly provided by Dr. Kapczynski, SEPRL, ARS, USDA.
h.
Millipore, Bedford, MA.
i.
Bethyl Laboratories Inc., Montgomery, AL.
j.
Instituto Rosenbusch S.A., Argentina.
k.
Laboratorios INMUNER, Argentina.
l.
Maxisorp, Nunc, Rochester, NY.
m.
Sigma-Aldrich, St. Louis, MO.
n.
Svanova Biotech AB, Uppsala, Sweden.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.