
Abstract
Polymerase chain reaction (PCR)-based assays combined with microchip electrophoresis were developed and evaluated for diagnosis and genotyping of GM2 gangliosidosis variant 0 (Sandhoff-like disease) in Japanese domestic cats. A preliminary genotyping survey was carried out in the population of Japanese domestic cats (1,015 cats in total) in southern Japan. Three kinds of assays including PCR primer-induced restriction analysis (PIRA) and mutagenically separated (MS)-PCR were carried out using blood-stained Flinders Technology Associates filter papers (FTA cards) as templates. The PCR products were analyzed by both agarose gel and microchip electrophoreses. All assays were sufficient to determine the genotypes of this disease, but MS-PCR offered the most rapid and simplest test, as it does not need the restriction enzyme step required in PCR-PIRA. The use of microchip electrophoresis in combination with FTA cards for sampling could shorten the time required for genotyping and simplify the procedure as well. The genotyping survey in the current study did not find any cats that possessed the mutant allele, suggesting that the prevalence of this allele is low (<0.1%) in southern Japan.
Keywords
The GM2 gangliosidoses are a rare group of lysosomal storage diseases resulting from excessive accumulation of GM2 ganglioside and related glycolipids in lysosomes, especially in the lysosomes of neurons, manifested as progressive neurological disorders involving motor and psychointellectual dysfunctions, visual defects, and other neurological disorders. 5 These diseases are inherited as autosomal recessive traits and caused by severely reduced activity of lysosomal β-N-acetylhexosaminidase (Hex), which hydrolyzes GM2 ganglioside. The lysosomal enzyme Hex consists of 2 subunits, α and β, which dimerize to form different isoforms of the enzyme, Hex A (αβ) and Hex B (ββ). Therefore, deleterious mutations in the HEXB gene encoding the β-subunit of Hex molecules affect both Hex A and Hex B, producing the null (0) variant of GM2 gangliosidosis (human Sandhoff disease) and lysosomal storage of undegraded GM2 ganglioside in neurons as a major storage material. 5
In domestic animals, naturally occurring Sandhoff-like disease has been reported in only 2 canine breeds (i.e., a Golden Retriever 18 and toy Poodles 16 ) and 4 feline breeds or families (i.e., domestic cats in the United States, 4 Korat cats, 13 Japanese domestic cats, 19 and European Burmese cats 3 ). At present, the pathogenic mutations in all 4 feline families have been identified, 3,8,11,12 but not in canine breeds. The mutation in Japanese domestic cats is a single nucleotide substitution from cytosine to thymine at nucleotide position 667 (c.667C> T) in the open reading frame (ORF) of the feline HEXB gene. 8 This mutant allele can be detected as an absence of digestion by the endonuclease RsaI in polymerase chain reaction (PCR) primer-induced restricted analysis (PIRA), which can be used to detect a single nucleotide mutation by introducing an artificial restriction endonuclease site using primers containing mismatches. 6 In this respect, the PCR-PIRA used to date is not completely specific for the mutant allele because the restriction site (GTAC) of RsaI is induced onto the wild allele by this method. In addition, recent reports suggest that this mutant allele is widely spread in the Japanese domestic cat population. 7,17 To the authors' knowledge, 9 affected cats have been diagnosed with Sandhoff-like disease over a wide geographic area from central (Kanto district) to southern (Kyushu district) Japan since the 1990s (O. Yamato, unpublished information, 2009). Therefore, it is likely that a veterinary practitioner will encounter affected cats within Japan and possibly abroad as well. Thus, simpler and more specific diagnostic assays are needed for such occasions. The present study developed and evaluated reliable, rapid, and simple PCR-based diagnostic assays for Sandhoff-like disease in Japanese domestic cats.
Whole blood samples were collected using heparin or ethylenediamine tetra-acetic acid (EDTA) as an anticoagulant from 6 wild-type, 5 heterozygous carrier, and 3 affected cats and stored them using Flinders Technology Associates filter paper (FTA card) a from a previous study. 7,8,17 The genotypes had been determined using a direct sequence analysis. 8 Heparinized whole blood samples were also collected from 1,015 mixed breed cats for screening. Each blood was spotted onto the FTA card, allowed to dry, and stored at approximately 4°C until used. A 1.2-mm diameter disc was punched out of the FTA card using a hole-punch a for each PCR. The disc was placed into a separate 0.2-ml tube for PCR. The disc on the tube bottom was washed 3 times for 5 min with 100 μl of a special washing solution, a rinsed twice for 5 min with 200 μl of Tris-EDTA buffer, b and dried at 60°C for 10 min. All PCR-based assays were performed using this treated disc as a template.
Characteristics of primers used in the current study.*
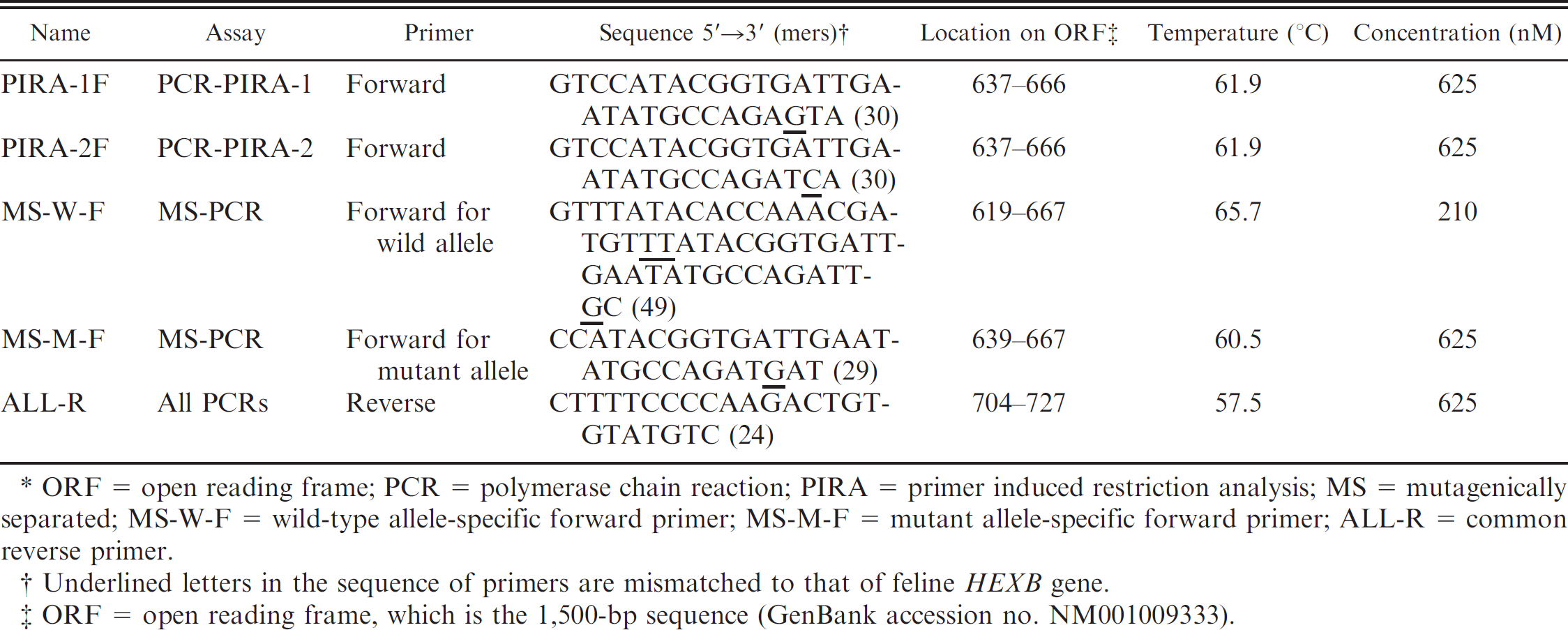
ORF = open reading frame; PCR = polymerase chain reaction; PIRA = primer induced restriction analysis; MS = mutagenically separated; MS-W-F = wild-type allele-specific forward primer; MS-M-F = mutant allele-specific forward primer; ALL-R = common reverse primer.
Underlined letters in the sequence of primers are mismatched to that of feline HEXB gene.
ORF = open reading frame, which is the 1,500-bp sequence (GenBank accession no. NM001009333).
Two types of PCR-PIRA (PCR-PIRA-1 and PCR-PIRA-2) were carried out using different forward primer and the same reverse primer (ALL-R) shown in Table 1. The PCR-PIRA-1 was a previously reported assay, 8 in which the 3′-end (GTA) of the forward primer (PIRA-1F) induces the restriction site (GTAC) of RsaI onto the wild allele as a result of combination with the wild genomic sequence (667C). In the present study, the PCR-PIRA-1 assay was carried out using an FTA card as a template. In the PCR-PIRA-2 assay, the 3′-end (CA) of the forward primer (PIRA-2F) induces the restriction site (CATG) of endonuclease NlaIII on the mutant allele as a result of combination with the mutant genomic sequence (667TG). The 2 PCR-PIRA assays were performed in 20 μl of reaction mixture containing 10 μl of 2× PCR master mix, c 12.5 pmol of each primer, and a treated disc of FTA card as a template. After the initial denaturation at 95°C for 2 min, 40 cycles of amplification were carried out, at a denaturing temperature of 95°C for 30 sec, an annealing temperature of 55°C for 30 sec, and an extension temperature of 72°C for 1 min. Extension during the last cycle was carried out at 72°C for 5 min. The product of the PCR-PIRA-1 assay was digested with RsaI d at 37°C for 90 min in 10 μl of reaction mixture containing 8 μl of PCR product, 15 units of RsaI, and 1 μl of 10× reaction buffer included by the manufacturer. The product of the PCR-PIRA-2 assay was digested with NlaIII e at 37°C for 90 min in 10 μl of reaction mixture containing 7 μl of PCR product, 10 units of NlaIII, 1 μl of 10× reaction buffer, and 0.1 μl of 100× bovine serum albumin included by the manufacturer.
The mutagenically separated (MS)-PCR assay was carried out according to the theory and rules reported previously. 14 The procedure was originally developed for easier genotyping of human familial hypercholesteremia in a single PCR tube without a restriction enzyme process. The design of primers, shown in Table 1, was based on the general rule of MS-PCR, 14 and the ratio of short-to-long allele-specific forward primers was increased to 3:1 on a molar basis (Table 1) to produce bands of similar intensity in the heterozygote when visualized on an agarose gel. In the MS-PCR, 2 allele-specific forward primers (MS-W-F and MS-M-F) and a reverse primer (ALL-R) were used simultaneously in a single PCR tube. The PCR was carried out using the same reagents and conditions as those in PCR-PIRA except for the annealing temperature (50°C).
All undigested and digested PCR products were subjected to electrophoresis in 3% (wt/vol) agarose gel d dissolved with Tris-EDTA buffer. b Molecular size markers d were used in the agarose gel electrophoresis. The analysis of PCR products was also performed using a microchip electrophoresis system f with its special reagent kit f including internal DNA size markers and DNA separation buffer. A fluorescent dye g was added to the DNA separation buffer according to the manufacturer's protocol of the microchip electrophoresis system. The PCR products were diluted 2–8 times with deionized water before application to this system to diminish the adverse influence of PCR and endonuclease buffers on electrophoretic mobility. The DNA ladder markers g from 25 to 450 bp were used as reference for DNA sizing.
Using the assays developed in the current study, an epidemiological survey was preliminarily carried out for the screening of the mutant allele in 1,015 mixed breed cats in Kagoshima, Japan. The cats were chosen from healthy and diseased patients in a Kagoshima animal hospital with the owners' informed consent from 2008 to date. The genotype of each cat was determined using 1 of the 3 assays described previously.
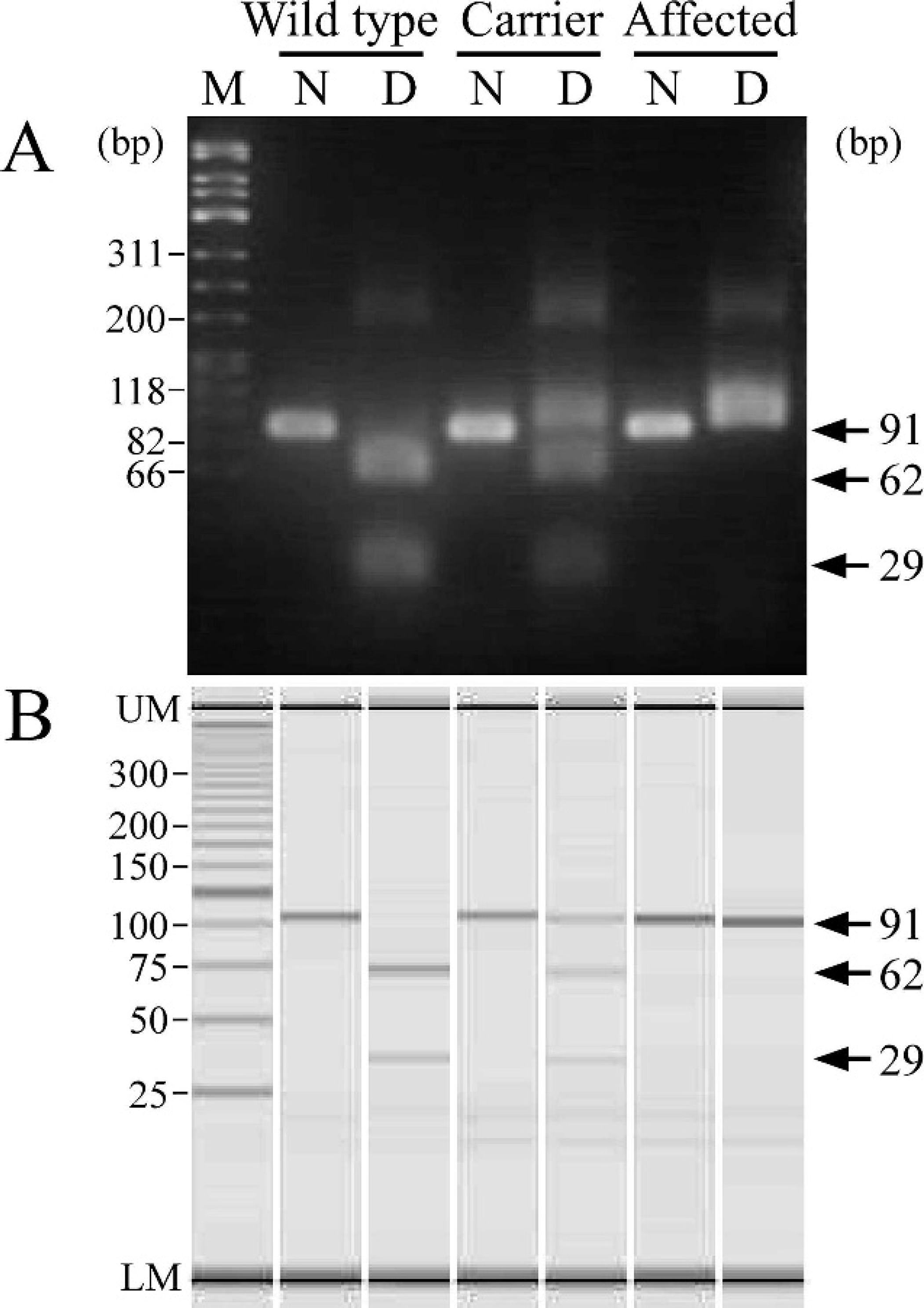
Genotyping of wild-type cats, heterozygous carriers, and affected cats from a family with Sandhoff-like disease by polymerase chain reaction primer-induced restriction analysis (PCR-PIRA)-1 using agarose gel (
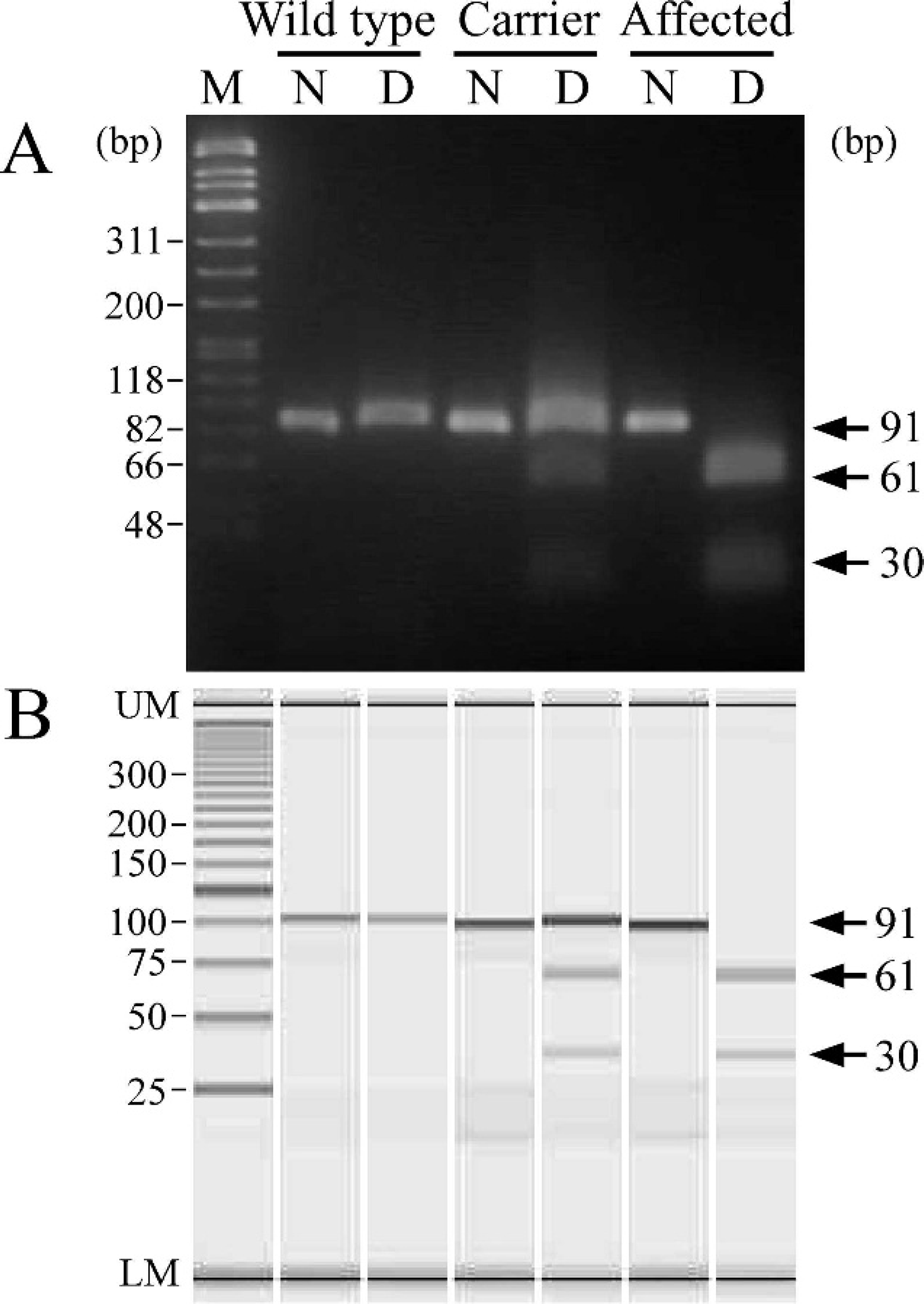
Genotyping of wild-type cats, heterozygous carriers, and affected cats from a family with Sandhoff-like disease by polymerase chain reaction primer-induced restriction analysis (PCR-PIRA)-2 using agarose gel (
As shown in Figure 1A, a 91-bp fragment was amplified by PCR-PIRA-1 and digested with RsaI, producing 62- and 29-bp bands in wild-type cats. However, a 91-bp fragment could not be digested with RsaI in affected cats, resulting in a band left on the gel after the digestion. The heterozygous carrier showed 1 undigested and 2 digested fragments on the gel. The 3 genotypes could be separated using the agarose gel electrophoresis, but the digested DNA bands on the gel tended to be broad and pale after the digestion, making it slightly difficult to identify them when a small amount of sample was applied to the gel. The results of microchip electrophoresis were identical to those achieved by gel electrophoresis (Fig. 1B). However, all DNA bands were sharp and clearly discriminated the 3 genotypes using the microchip electrophoresis system, making it easy to read the result.
Figure 2 shows the results of the PCR-PIRA-2 assay. The PCR-PIRA-2 assay had 1 band in wild-type cats and two 61- and 30-bp bands in affected cats after the digestion of a 91-bp fragment with NlaIII, which was different from the PCR-PIRA-1 assay. In the PCR-PIRA-2 assay, the heterozygous carrier showed both digested and undigested fragments on the gel, just like in the PCR-PIRA-1. In addition, the microchip electrophoresis yielded sharp and clear bands, making it easy to discriminate the 3 genotypes.
As expected, in the MS-PCR, a 109-bp fragment was amplified with a wild allele-specific forward primer and a common reverse primer in wild-type cats, and an 89-bp fragment with a mutant-specific forward primer and the common reverse primer in affected cats (Fig. 3A). In heterozygous carriers, these 2 bands of similar intensity were clearly seen on the gel. The result obtained using microchip electrophoresis was consistent with that of the agarose gel electrophoresis (Fig. 3B).
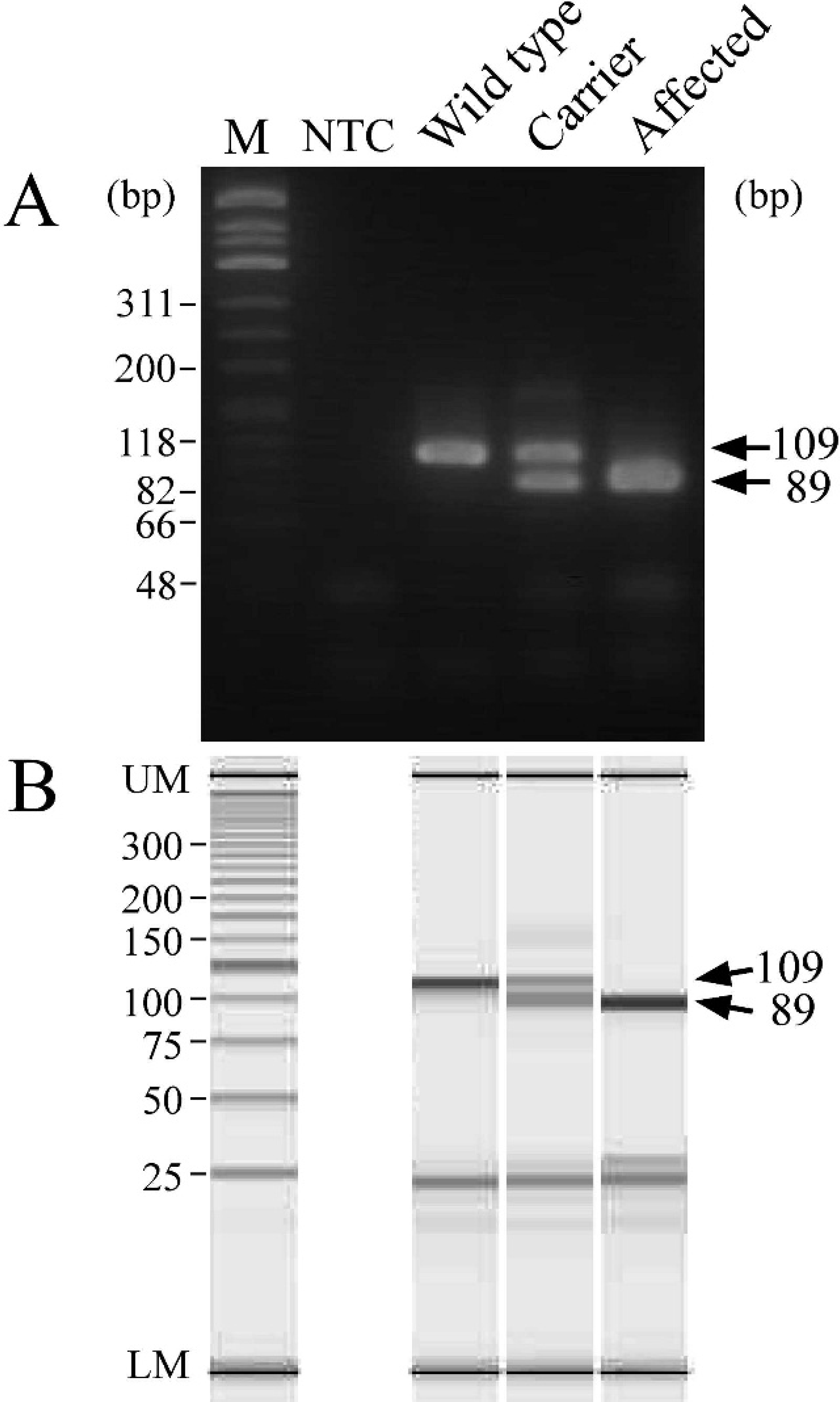
Genotyping of wild-type cats, heterozygous carriers, and affected cats from a family with Sandhoff-like disease by mutagenically separated polymerase chain reaction using agarose gel (
All cats examined in the present study were wild type in genotypes. Neither carrier nor affected genotypes were found in this cat population. All PCR-based assays used in the present study (i.e., PCR-PIRA-1, PCR-PIRA-2, and MS-PCR) were almost sufficient to determine the genotypes of Sandhoff-like disease in Japanese domestic cats (Figs. 1–3). The use of FTA cards for sampling shortened the time required for genotyping and simplified the procedure because blood-stained FTA cards can be stored stably for a long time even at room temperature and used conveniently as a direct template for PCR after a simple pretreatment. However, caution should be used when different conditions are used for PCR-PIRA-1 in combination with a treated disc from an FTA card. Previously, in the PCR-PIRA-1 assay, a different Taq polymerase was used, and FTA cards were not used. 8,17 The PCR-PIRA-1 assay was combined with FTA cards in the study. In the present study, the PCR-PIRA-1 assay was initially carried out using the previously used Taq polymerase and a pretreated disc of FTA card as a template, but a 91-bp fragment derived from wild-type cats did not completely disappear after digestion with RsaI for unknown reasons (data not shown), and made it impossible to clearly distinguish a wild genotype from a heterozygous genotype. In addition, RsaI in the PCR-PIRA-1 assay does not digest unknown mutations adjacent to nucleotide position 667 in the ORF of feline HEXB gene. Therefore, although the PCR-PIRA-1 assay is not specific to c.667C> T, the PCR-PIRA-2 assay is specific to c.667C>T because NlaIII cleave only the mutant sequence.
In the present study a single-step assay was also proposed using MS-PCR theory for genotyping of feline Sandhoff-like disease. In principle, MS-PCR offers a rapid, simple, and inexpensive test as it does not require a restriction enzyme process, and the procedure can be used for genotyping several inherited diseases or genetic traits, 9,10,14,15 although it is rather difficult to design appropriate primers and find the optimal PCR conditions. A suitable method was finally established by a process of trial and error, and therefore all reagents and conditions for this PCR should not be changed to prevent false results. Consequently, the successful result in the current study (Fig. 3) verified that MS-PCR is the simplest, most rapid, and least expensive assay for the genotyping of feline Sandhoff-like disease provided that blood-stained FTA cards are used as templates.
Microchip electrophoresis has recently attracted considerable attention in DNA analysis due to its high efficiency, high throughput, time-saving ability, easy operation, and low consumption of samples and reagents. 1,20 Furthermore, gel electrophoresis occasionally requires hazardous substances (acrylamide gel) or mutagenic (ethidium bromide) and toxic (silver nitrate) stains, whereas the use of toxic substances and expenses incurred for hazardous waste disposal can be avoided using microchip electrophoresis. 2 Therefore, in the present study, the combination of the PCR-PIRA and MS-PCR protocols with the microchip electrophoresis system was attempted to find an approach with the highest potential for the genotyping procedure. As a result, analysis by microchip electrophoresis clearly exhibited all DNA bands that are essential for genotyping in both PCR-PIRA (Figs. 1B, 2B) and MS-PCR (Fig. 3B). In addition, the use of microchip electrophoresis in combination with FTA cards and the MS-PCR can markedly shorten the time for analysis of DNA fragment patterns, which is approximately 3 min per sample, allowing for early reporting of results within 4 hr after sample collection.
The survey in the present study suggested that the allele frequency of feline Sandhoff-like disease is low (<0.1%) in the mixed breed cat population in southern Japan. However, further study is needed to clarify the frequency and distribution of this mutant allele in Japan and other countries.
In conclusion, the present study demonstrates that MS-PCR coupled with microchip electrophoresis system is a suitable method of determining the genotypes of Sandhoff-like disease in Japanese domestic cats among PCR-based assays used in the study. In addition, this technique, in combination with FTA cards for sampling, can further shorten the time and simplify the procedures. Therefore, these techniques can be particularly useful in the diagnosis and screening of Sandhoff-like disease.
Acknowledgements
The study was supported financially by grants (20380173, 20–08112, and 21658109, OY) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) and by the Strategic Research Base Development Program for Private Universities, 2008–2012 (TA), which is also funded by MEXT.
Footnotes
a.
FTA classic card, Harris Uni-Core Punch (size 1.2 mm), FTA Purification Reagent, Whatman International Ltd., Piscataway, NJ.
b.
zTris-EDTA buffer, pH 8.0, Wako Pure Chemical Industries Ltd., Osaka, Japan.
c.
GoTaq Green Master Mix (2×), Promega Corp., Madison, WI.
d.
RsaI, Agarose 21, OneSTEP Marker 9, Nippon Gene Co. Ltd., Tokyo, Japan.
e.
NlaIII, New England Biolabs Inc., Ipswich, MA.
f.
MCE-202 MultiNa, DNA-500, Shimadzu Corp., Kyoto, Japan.
g.
SYBR Gold Nucleic Acid Gel Stain, 25-bp DNA ladder, Invitrogen Corp., Carlsbad, CA.