
Abstract
The bacterium Coxiella burnetii, which has a wide host range, causes Q fever. Infection with C. burnetii can cause abortions, stillbirth, and the delivery of weak offspring in ruminants. Coxiella burnetii infection is zoonotic, and in human beings it can cause chronic, potentially fatal disease. Real-time polymerase chain reaction (PCR) is increasingly being used to detect the organism and to aid in diagnosis both in human and animal cases. Many different real-time PCR methods, which target different genes, have been described. To assess the comparability of the C. burnetii real-time PCR assays in use in different European laboratories, a panel of nucleic acid extracts was dispatched to 7 separate testing centers. The testing centers included laboratories from both human and animal health agencies. Each laboratory tested the samples using their in-house real-time PCR methods. The results of this comparison show that the most common target gene for real-time PCR assays is the IS1111 repeat element that is present in multiple copies in the C. burnetii genome. Many laboratories also use additional real-time PCR tests that target single-copy genes. The results of the current study demonstrate that the assays in use in the different laboratories are comparable, with general agreement of results for the panel of samples.
With the exception of Antarctica and New Zealand, Q fever is a worldwide endemic disease. The causative agent of Q fever is Coxiella burnetii, a small Gram-negative obligate intracellular bacterium. 1 Ruminants are the most common animal reservoir for the bacterium. 3 Infection of ruminants is usually subclinical, but clinical signs including abortions, stillbirths, and the delivery of weak offspring can occur. 1
Infected animals can excrete C. burnetii in feces, urine, milk, and birth products. The bacterium can survive extracellularly in the environment for extended periods of time (up to 150 days) while still remaining infectious1 and has been reported 19 to have an infectious dose as low as a single bacterium. Naive animals become infected either through the inhalation of contaminated aerosols or through the ingestion of infectious matter. 3
A zoonotic disease, Q fever may develop into potentially fatal chronic disease in human beings. 1,20 Coxiella burnetii infection in humans can be associated with an outbreak or can be acquired sporadically through occupational exposure to animals or animal products, thus presenting an increased risk of disease. 13 Outbreaks of Q fever in human beings are often linked to airborne spread of the disease from infected ruminants, particularly during parturition. 8,9,14,21,23 As a result of its high infectivity, the possibility of spread via aerosols, and the potentially fatal consequences of infection, C. burnetii is categorized as a biosafety level 3 agent.
Over recent years, there have been several high-profile Q fever outbreaks in Europe, including a large outbreak that is currently ongoing in The Netherlands. 9,18,22 The increasing profile of the disease has led to renewed interest in the diagnosis of cases of Q fever both in human beings and animals, and real-time polymerase chain reaction (PCR) is rapidly being adopted throughout Europe to aid in the detection of C. burnetii.
A ring-trial was arranged involving 7 European testing centers, comprising both veterinary and human laboratories, to compare a number of the real-time PCR methods currently in use. Seven laboratories participated in the ring-trial, including the following: Veterinary Laboratories Agency (VLA), Weybridge, United Kingdom; Health Protection Agency, Porton Down, United Kingdom; Royal Hospitals, Belfast, United Kingdom; National Institute for Public Health and the Environment, Bilthoven, The Netherlands; Federal Institute for Risk Assessment, Berlin, Germany; National Veterinary Institute, Uppsala, Sweden; and Central Veterinary Institute of Wageningen UR, The Netherlands.
As a result of the designation of C. burnetii as a biosafety level 3 agent, there are concomitant difficulties with transporting positive sample material to laboratories located around Europe. Therefore, nucleic acid extracts were prepared from the sample panel at a central laboratory (VLA) and dispatched frozen to the participating testing centers. Samples were of caprine, ovine, and bovine origin and consisted of placental cotyledons or fetal fluid samples from abortion cases. Samples that were designated as “test-positive” had previously been demonstrated to contain C. burnetii through the use of immunohistochemistry or through microscopic evaluation of stained smears. Prior to dispatch, the suitability of the sample panel for the present study was assessed using a real-time PCR assay targeting the IS1111 repeat element.
A small section of placental cotyledon tissue (1–10 mg) was mechanically disrupted using a micropestle and mortar. Both the tissue samples and the fetal fluid samples were subjected to an external overnight lysis. The DNA was then extracted from the digested samples using an extraction robot a along with the dedicated buffers contained in the associated reagent kit. a Samples were diluted in nuclease-free water if required.
Sample panel composition.
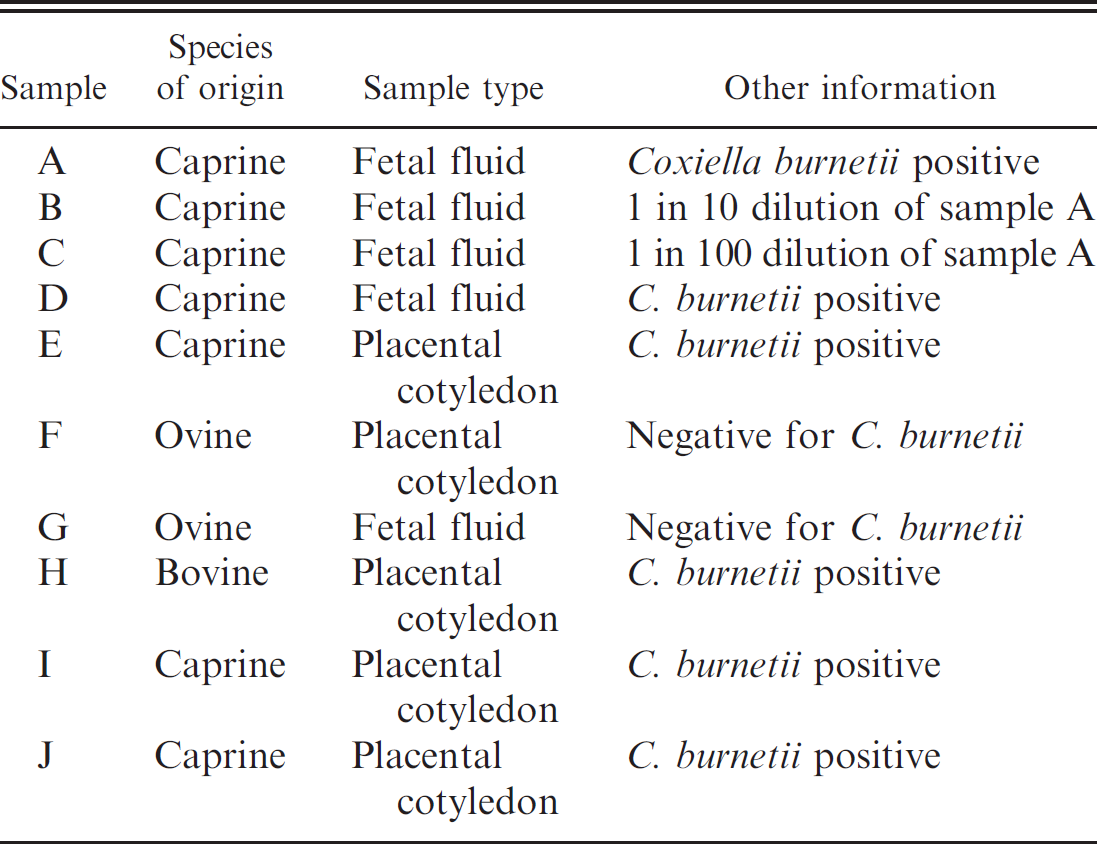
Each testing center received a panel of 10 nucleic acid samples, which included extracts from 2 samples that had previously tested negative for C. burnetii and 6 samples that had previously tested positive for C. burnetii. To assist in distinguishing between the efficacies of different real-time PCR methods, 1 sample (sample A) was subjected to a 10-fold serial dilution to produce samples B and C. The composition of the sample panel is shown in Table 1.
Each participating laboratory tested the panel of nucleic acid extracts using their in-house assay(s) (Table 2). Testing centers were blinded to the C. burnetii status of the samples being tested. In some laboratories, the samples were tested using more than one real-time PCR, and at some testing centers, the samples were tested by multiple operators. Each real-time PCR assay was used to test the nucleic acid samples using between 2 and 6 replicate reactions for each template.
The results of the ring-trial (Table 2) demonstrated that samples A, D, E, H, I, and J were positive for C. burnetii with all real-time PCR methods used. Samples F and G had been included as samples, which had previously tested negative for C. burnetii. Sample G tested negative with all of the real-time PCR assays used, while sample F was negative in the majority of assays, with 1 assay showing amplification in a single reaction.
The inclusion of samples B and C allowed for discrimination between the differing sensitivities of the assays (Table 2). Sample B was positive in all of the assays targeting the IS1111 repeat element, while only 1 single-copy gene assay was able to detect the C. burnetii DNA present in this sample. Coxiella burnetii DNA was not detected in sample C by any of the PCR assays targeting single-copy genes, and C. burnetii DNA could only be detected in sample C using 2 of the 7 IS1111 real-time PCR assays.
Strikingly, when the data for each positive sample are examined at the level of quantification cycle, there is close agreement between the results obtained with the real-time PCR methods that target the same region of the C. burnetii genome. This is especially surprising given that different PCR reagents and real-time PCR platforms were used in the different testing centers. Additionally, when each assay is used to rank the samples in order of increasing bacterial load, there is again close agreement among the assays.
Comparison of real-time polymerase chain reaction results generated from 7 participating laboratories. *
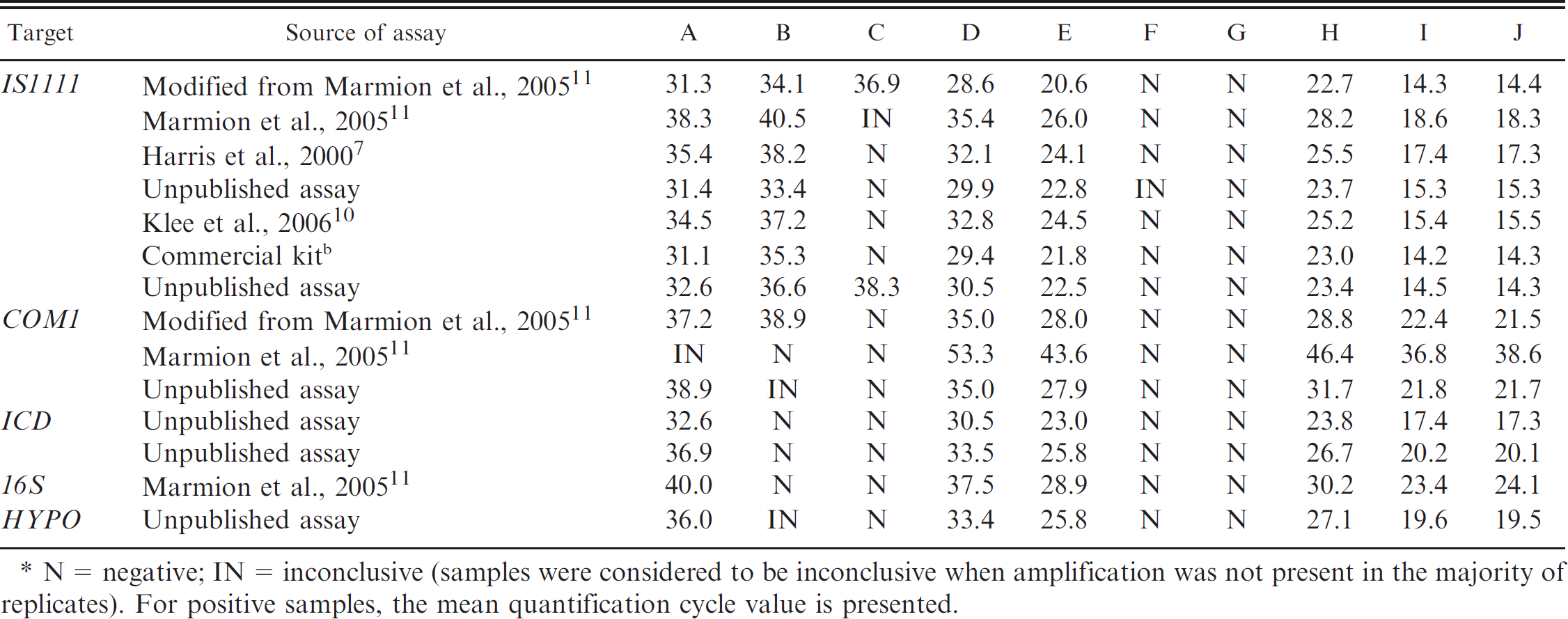
N = negative; IN = inconclusive (samples were considered to be inconclusive when amplification was not present in the majority of replicates). For positive samples, the mean quantification cycle value is presented.
The present survey of the real-time PCR assays used to detect C. burnetii in different European laboratories showed that all of the participating laboratories were using assays targeting the multicopy IS1111 repeat element. Many laboratories used multiple real-time PCR assays, combining an IS1111 assay with assays targeting single-copy genes such as COM1 or ICD.
Disease outbreak investigations may cross borders, involving agencies in different countries, and may involve both human and animal health agencies within a country. Given this, it is important that comparability is achieved for C. burnetii detection among these different agencies. The results of the current study indicate that the real-time PCR methods used to detect C. burnetii are comparable in different testing centers across the European Union and that laboratory test result agreement exists between animal and human agencies within the same country.
The results of the current study indicate that real-time PCR assays that target multiple copy genes, such as the IS1111 gene, are more sensitive than are those targeting single-copy genes, such as the COM1 or ICD genes. However, many laboratories use additional assays, such as those targeting single-copy genes. This provides an additional level of certainty for any positive result, as it demonstrates that multiple independent sections of the C. burnetii genome can be detected in a sample. Additionally, assays targeting the IS1111 repeat element cannot be used for accurate quantification of the load of C. burnetii present within a sample, as this element is present in differing numbers (7–110) 10 depending on the strain of the bacterium present in the sample. The additional use of a PCR targeting a single-copy gene might allow accurate quantification of the bacterial load present within a sample. It is also worth noting that the single-copy methods were able to accurately show the presence of C. burnetii DNA in typical placental cotyledon and fetal fluid samples, such as those used in the current exercise.
Use of real-time PCR provides a sensitive method of detecting C. burnetii in animal and human samples. Unlike serological assays, PCR can be used to demonstrate the presence of the organism within clinical material, 2,4–6,12,15,16 and, by doing so, it provides important information to the veterinarian, which will help to inform the final diagnosis. This may be especially relevant in cases involving ruminant abortions, in which demonstrating the presence of C. burnetii within the abortion material may implicate the organism as the cause of the abortion. Critically, subject to a suitable extraction method, real-time PCR might be used to detect the presence of C. burnetii within a multitude of different sample types, including bedding material, straw, and birth products (including placental and fetal material), that could be associated with the production of contaminated aerosols, which may be potentially involved in a disease outbreak. 9,17,23 Polymerase chain reaction assays may also be used to detect C. burnetii in milk. 15 The current study has shown that suitable detection methods are available in many laboratories to detect C. burnetii in a disease outbreak.
Acknowledgements. The authors would like to thank Jane Errington and Sue Hannon for performing the nucleic acid extractions and the staff from the Veterinary Laboratories Agency Regional Laboratories for providing samples for use in the study. This work was supported by the FZ2100 non-statutory zoonoses surveillance budget, the VLA Test Development Programme, and by the MedVetNet Workpackage 25. British Crown copyright and Crown user rights are reserved.
Footnotes
a.
MagNA Pure LC, Roche Diagnostics Ltd., Burgess Hill, United Kingdom.
b.
Adiavet Cox, Adiagene, Saint Brieuc, France.