
Abstract
The use of embryonating chicken eggs in preparation of avian virus vaccines is the principle cause for contamination with Chicken anemia virus (CAV). Identification of CAV in contaminated vaccines relies on the expensive, tedious, and time-consuming practice of virus isolation in lymphoblastoid cell lines. The experience of the last 2 decades indicates that polymerase chain reaction is extending to replace most of the classic methods for detection of infectious agents. In the present report, a simple, rapid, and accurate polymerase chain reaction method for detection of CAV in poultry vaccines is described. Oligonucleotide primers homologous to highly conserved sequences of the VP1 gene were used to amplify a fragment of 676 bp. The developed assay was specific for detecting CAV from different sources, with no cross reactivity with many avian viruses. No inter- and intra-assay variations were observed. The analytical sensitivity of the test was high enough to detect 5 TCID50 (50% tissue culture infective dose) of the virus per reaction; however, different factors related to the vaccine matrix showed considerable effects on the detection limit. In conclusion, this method may represent a suitable alternative to virus isolation for identification of CAV contamination of poultry virus vaccines.
Introduction
Chicken anemia virus (CAV) is a small, non-enveloped, icosahedral virus that contains a circular, single, negative-strand DNA genome. 23 It is the causative agent of chicken infectious anemia (CIA) and classified as the sole member of genus Gyrovirus within the family Circoviridae. 19 The clinical picture of CIA often appears in young, 2–3-week-old chicks, which usually acquire the infection vertically, and it is manifested by aplastic anemia and death. 14 Older chickens, which become infected horizontally when maternally derived antibody fails to be protective, 14 only develop subclinical infection evidenced by poor response to vaccines and increased susceptibility to other infections. 25
Since the first isolation of CAV from contaminated vaccines in Japan in 1979, 33 the role of CAV as a vaccine contaminant is well established. The virus usually introduces to the avian vaccines prepared in embryonating eggs, which get the infection vertically. However, the use of specific pathogen–free (SPF) eggs did not eliminate the possibility of vaccine contamination. Previous studies 4,5 reported evidence about the persistence of CAV in the gonads and its vertical transmission to the progeny of SPF chickens. The presence of viral DNA in a large percentage of blastodisks, collected from freshly laid SPF eggs and in the embryos at 16–19 days of incubation further potentiates this evidence. 16
National and international vaccine production regulations 6,7,18,26 require the screening of avian vaccines for a multitude of extraneous viruses, including: Newcastle disease virus, Avian leukosis virus, Avian reticuloendotheliosis virus, Avian reovirus, Gallid herpesvirus 1, Infectious bursal disease virus, Infectious bronchitis virus, and CAV. Assays described for detection of such contaminants often involve virus isolation in cell culture, embryonating eggs, or laboratory animals. Although this approach is established and routinely used, it is laborious, expensive, and time consuming. 12 In addition, several virologic and serologic tests are used, including hemagglutination/hemadsorption, immunofluorescence and enzyme-linked immunosorbent assay (ELISA), but these methods usually have variable degrees of limited specificity, sensitivity, and applicability. Well-designed polymerase chain reaction (PCR) assays provide a faster and inexpensive alternative to those techniques, and its established higher sensitivity and specificity has been documented. 1
Avian viral vaccines used in specificity testing and spiking experiments.
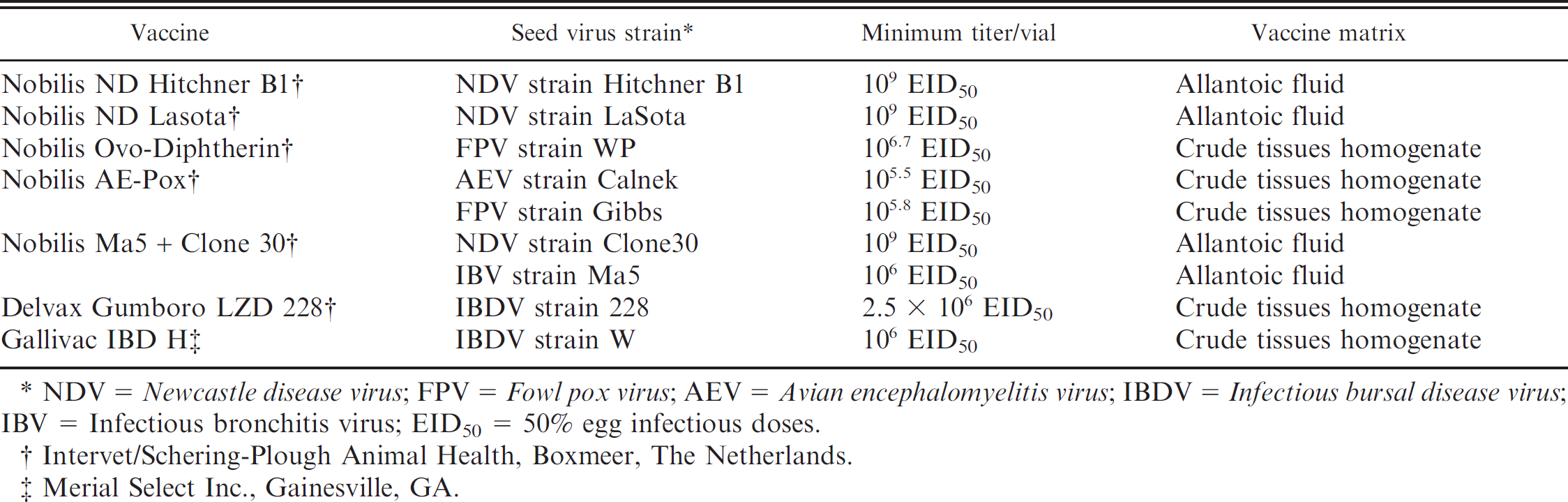
NDV = Newcastle disease virus; FPV = Fowl pox virus; AEV = Avian encephalomyelitis virus; IBDV = Infectious bursal disease virus; IBV = Infectious bronchitis virus; EID50 = 50% egg infectious doses.
Intervet/Schering-Plough Animal Health, Boxmeer, The Netherlands.
Merial Select Inc., Gainesville, GA.
Different PCR formats have been developed for detection and quantification of CAV DNA, including endpoint PCR, 10,17 competition quantitative PCR, 31 strain-specific real-time PCR, 13 and real-time quantitative PCR–based serum neutralization test. 27 None of these assays were validated for use in the detection of CAV as an extraneous virus agent in poultry vaccines. Validation of PCR assays for use in the detection of CAV in different viral vaccine formulations is essential for 2 reasons. First, constituents added during vaccine formulation may interfere with virus extraction and/or PCR amplification. Second, DNA from virus propagation hosts may produce false-positive or confusing PCR results. Therefore, the current study was designed for developing and optimizing a high-performance PCR assay to detect CAV in live avian virus vaccines. Different aspects were validated, including the assay's specificity, sensitivity, reproducibility, and effect of the vaccine matrix.
Materials and methods
Vaccines
A commercially available live CAV vaccine a that contains 106 TCID50 (50% tissue culture infective dose) of strain 26P4a was kindly supplied by the Central Laboratories for Control of Veterinary Biologics, Abassia, Egypt, for use in the development of the CAV PCR assay. The same virus was used for spiking different dilutions of avian viral vaccines to assess the utility of the CAV PCR assay in the detection of CAV contamination of vaccines. Seven different avian live virus vaccines, prepared on chicken embryos and proved to be free from CAV contamination, were used as vehicles for spiking experiments and for testing the developed assay's specificity (Table 1).
Oligonucleotide primers. *

CAV = Chicken anemia virus; PCR = polymerase chain reaction.
Positions relevant to CAV isolate 01-4201 (accession no. DQ991394.1).
Sensitivity and spiking
Ten-fold serial dilutions, from 10−1 to 10−8, of the rehydrated CAV vaccine were prepared in PCR-grade water as well as in the different rehydrated live avian virus vaccines (Table 1). The DNA extracts from the stock as well as each dilution were tested by using the PCR assay described in the following section. Serial 2-fold dilutions were prepared from the lowest dilution giving specific amplification in the initial testing. Extracts from the 2-fold serial dilutions were extracted and tested as the initial 10fold dilutions.
Oligonucleotide primers
A wide range of CAV sequences available on the genetic sequence databases (accession nos. FJ172347.1, AY846844.1, DQ217400.1, DQ217401.1, AF311900.3, DQ991394.1, EF176599.1, DQ141672.1, DQ141673.1, AF390038.1, AJ297685.2, AJ297684.2, AB119448.1, and AY040632.1) were collected and aligned by using the MegAlign program, version 3.18. b Two previously published oligonucleotide primers 24,28 that detected highly conserved sequences within the CAV VP1 gene (Table 2) were selected. Their specificity was confirmed by Primer-BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), and suitability was verified by analysis that used Oligo design and analysis tools. c The primers were synthesized by Metabion. d
DNA extraction
All vaccines were first resuspended in 1 ml of sterile phosphate buffered saline solution (pH 7.4). The DNA extraction of the vaccine preparations and all the spiked dilutions were conducted by using a commercial DNA extraction kit. e The procedure was carried out by using a 200-μl sample volume according to the manufacturer's protocol. Purified DNA was recovered in a 50-μl elution buffer and stored at −20°C until use.
Sensitivity of Chicken anemia virus polymerase chain reaction (PCR) assay in commercial poultry vaccines.
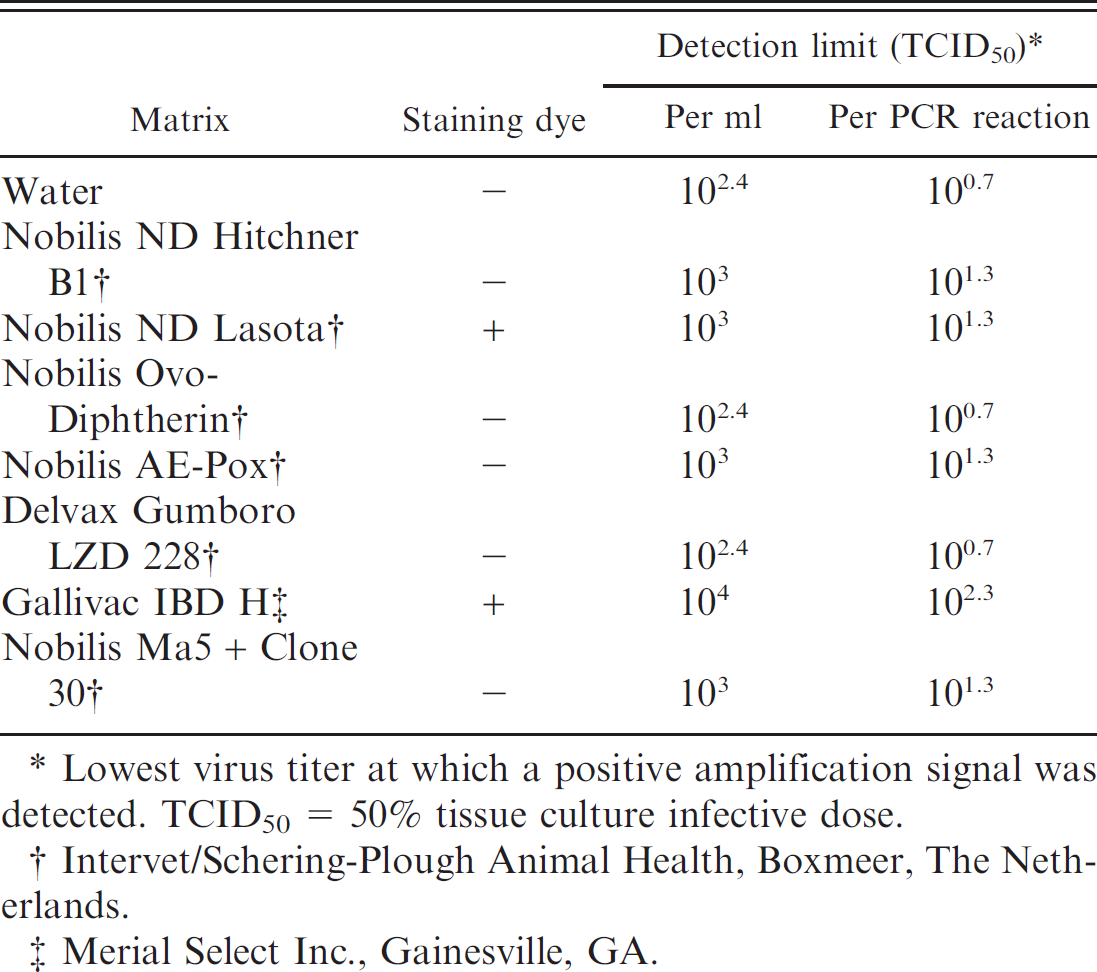
Lowest virus titer at which a positive amplification signal was detected. TCID50 = 50% tissue culture infective dose.
Intervet/Schering-Plough Animal Health, Boxmeer, The Netherlands.
Merial Select Inc., Gainesville, GA.
PCR amplification and gel electrophoresis
A reaction mixture, which contained 12.5 μl of 2× ReddyMix PCR Master Mix, f 1 μM (2.5 μl) of each primer, and 5 μl of DNA extract, was brought up to a 25-μl volume by using PCR-grade water. Target DNA sequences were amplified in 0.2-ml thin-walled tubes incubated in a thermal cycler. g The thermal profile of DNA amplification consisted of 1 cycle at 95°C for 5 min followed by 50 cycles of 95°C for 30 sec, 55°C for 35 sec, and 72°C for 1 min, and finally 1 cycle of 72°C for 7 min. Ten microliters of the PCR products were separated in 1.2% agarose gel that contained 0.5 μg/ml ethidium bromide. The specific bands were identified in comparison with 100-bp DNA ladder f and documented by using an image analysis system. h
Assay specificity and precision
The assay specificity was evaluated by testing preparations that contained CAV against DNA extracted from the different avian live virus vaccines (Table 1) for cross reactivity. To assess the assay's precision (reproducibility), 3 separate dilution series were assayed in a single run to determine the intra-assay variation. Interassay variation was also determined by application of 3 separate runs on different days by using freshly prepared reagents.
Results
Optimization of the PCR conditions
Different variables of the PCR assay were evaluated to identify the best possible conditions for getting highly specific and reproducible results. Three integrated kit systems were assessed and compared, including Illustra PuReTaq Ready-To-Go PCR beads, i ReddyMix PCR Master Mix, f and GeneAmp Fast PCR Master Mix. g All 3 systems were efficient in amplifying the target 676-bp fragment of CAV genome as visualized in agarose gel; with a slightly higher performance by ReddyMix and GeneAmp. In addition, other parameters such as magnesium chloride (MgCl2) and primer concentrations, annealing temperature, and cycling repeats were tested in series to establish the optimal working conditions. ReddyMix was selected to complete the PCR for economic reasons.
Specificity of the reaction
The specificity of PCR was verified by successful amplification of the target sequence in several batches of the CAV vaccine. Furthermore, no specific amplification was detected either with the negative control or with any other tested avian viruses, including Newcastle disease virus, Infectious bursal disease virus, Infectious bronchitis virus, Fowl pox virus, and Avian encephalomyelitis virus.
Detection limit and effect of different vaccine matrices
The sensitivity of the developed assay for detecting possible CAV contamination of commercial poultry vaccines was evaluated by testing 10-fold dilution series of the virus stock (106 TCID50/ml; dilutions 10−1 to 1018). Further 2-fold serial dilutions were applied to the lowest dilution that generated a positive signal. The results showed that the assay detected down to the genome equivalent of 250 TCID50/ml (the genome equivalent of 5 TCID50 per reaction; Table 3; Fig. 1). In certain cases, vaccine matrix reduced the assay performance and decreased the detection sensitivity 4–40-fold (Table 3; Fig. 1).
Reproducibility
All the replicates showed specific amplification of the target. Differences were observed only in band intensity in ethidium bromide gels.
Discussion
The use of high-quality vaccines is essential for maintenance of animal health and production. Different requirements and procedures were established for assuring the production of a proper vaccine for immunization purposes as described in the Code of Federal Regulations (CFR Title 9, 113), 26 in the annex of European Union Directive 2001/82/EC, 7 in the European pharmacopeia, 6 and in the World Organization for Animal Health manual of diagnostic tests and vaccines. 30 Freedom of the vaccine from contamination by extraneous virus agents should be secured throughout the process of production and before market release to avoid infection of immunized animals. 30
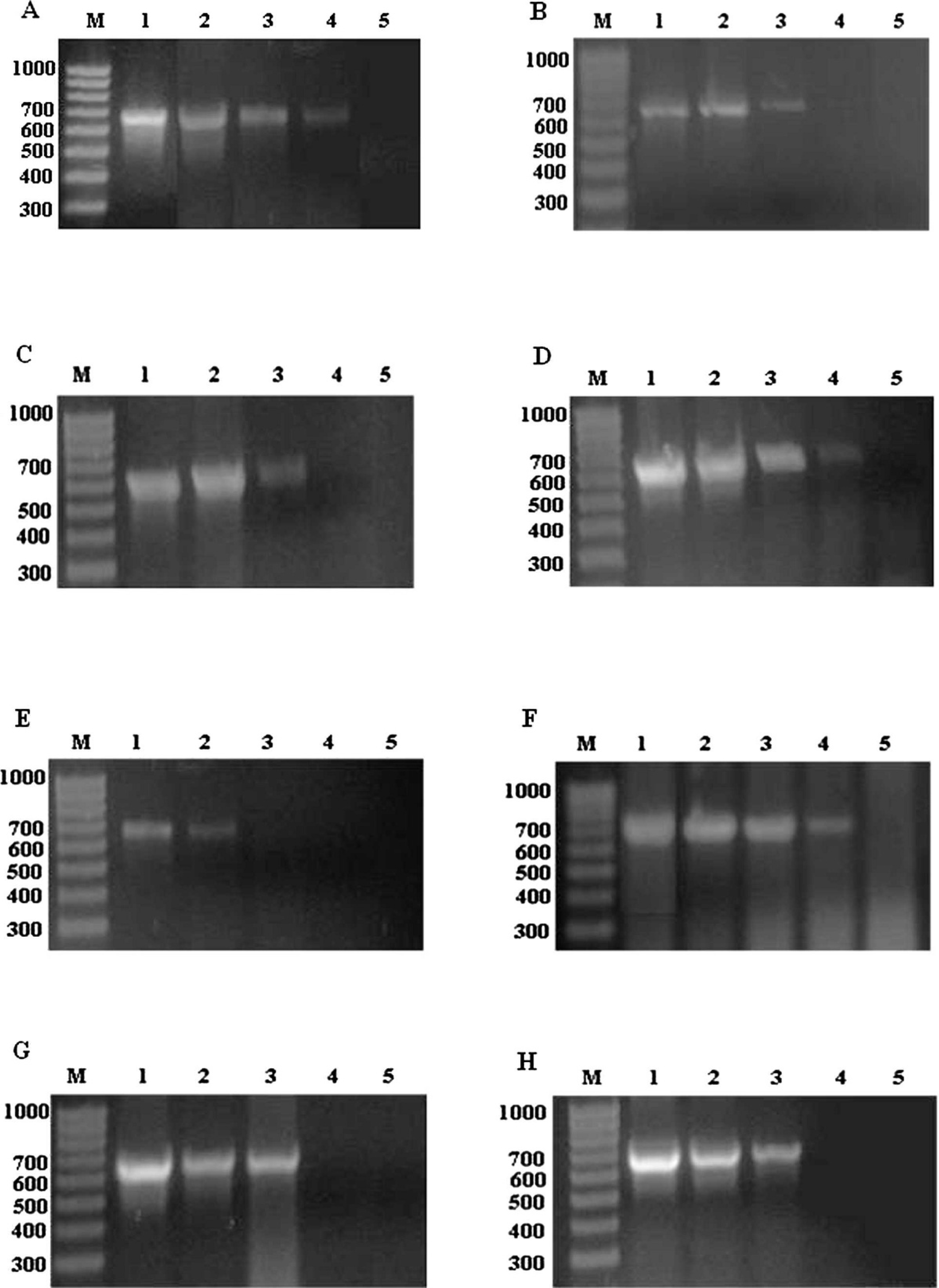
Polymerase chain reaction (PCR) detection of dilutions of Chicken anemia virus (CAV) in different matrices. PCR-grade water (
Chicken anemia virus is an important extraneous pathogen that potentially contaminates the avian virus vaccines, particularly those prepared by inoculation on chicken embryos. The virus was reported to infect SPF flocks, which supply eggs for vaccine production, and its DNA was detected in a large proportion of the laid eggs. 4,5,16 Vaccine contamination with CAV either results in severe anemia and significant mortalities in vaccinated birds or in potential reduction in the immune response to the target vaccine candidate(s). 25 Therefore, the process of assuring freedom of the avian virus vaccines from CAV contamination requires a 2-step strategy; first, testing the SPF embryonating eggs used in vaccine production and quality control; and second, examining the produced vaccines for possible contamination before release. In the current report, the development of a reliable and accurate assay for routine identification of CAV in poultry vaccines was described, whereas the establishment of a similar method for screening SPF eggs is a matter of ongoing research.
To date, identification of CAV as a possible vaccine contaminant is routinely done by virus isolation on cell culture, embryonated eggs, or SPF chickens. The MDCC-CU147 or MSB1 cell cultures are the most common hosts used, although some virus strains do not grow well and differences in susceptibility of MSB1 sublines have been reported. 18 Moreover, the traditional virus isolation methods relied on difficult, time-consuming passaging of cells until the culture fails because of CAV infection. 32 The PCR assay described in the current study, however, is a simple, rapid, and more accurate method for detecting several extraneous virus agents, such as Newcastle disease virus, 21 Infectious bronchitis virus, 8 Avian leukosis virus, 9 Infectious laryngotracheitis virus, 29 and Avian reovirus. 3 Hence, the proven aptitude of PCR was exploited in the current study to provide a suitable alternative to virus isolation for detecting CAV in vaccine preparations.
Several CAV genetic groups have been recognized, 11 with no significant antigenic or pathogenic differences among the isolates. 15 Therefore, the primers used in developing the current PCR assay were chosen to target highly conserved regions of the CAV genome after sequence alignment of 14 CAV strains.
During the primer selection phase, it was noticed that the published primers were not all suited for vaccine screening applications. The primer sequences published for the detection of CAV in formalin-fixed tissues did not yield any positive results when tested against published GenBank sequences. 10 Primers based on VP3 were not considered in our selection because they had an improved sensitivity only when applied as a nested procedure that is laborious and time consuming. 2 In comparison, the VP1 primers CAV1 and CAV2 hybridize to highly conserved sequences; thus ensuring that all variants of the CAV will be detected and that future mutations of the virus will not affect our primer binding and/or detection sensitivity. 24 It is important to note that CAV1 and CAV2 have never been tested for sensitivity of detection under any experimental conditions before the current work.
Specificity of the reaction was proven by testing several CAV vaccine batches, all of which produced a distinct amplification signal revealed by a band of 676 bp in agarose gel. Furthermore, no specific product was obtained with the no template control (NTC) and with other avian viruses such as Newcastle disease virus, Infectious bronchitis virus, Avian erythroblastosis virus, Infectious bursal disease virus, and Avian papillomavirus. The assay performance was further optimized by testing several variables of the reaction components and the cycling conditions (data not shown). This enabled consistent production of the specific PCR product in all reaction tubes, with no effect of the intra-assay or interassay factors.
The detection limit of the test (100.7 TCID50 per reaction; Table 3) is comparable with, or below, the detection thresholds of most developed PCR assays for CAV. 10,20,22,24 Although the use of nested PCR showed improved detection sensitivity by 10–100-fold, 10,20 it increases the cost and time required for vaccine evaluation with no extra positive impact on the results obtained.
Spiking of CAV in different commercial avian virus vaccines enabled the study of the effect of vaccine preparation method and vaccine matrix on the efficiency of the developed assay. In contrast to previous observations, 3 CAV was detected in vaccines produced in allantoic fluid such as Newcastle disease virus with the same efficiency that it was recovered from the vaccines produced as a crude tissues homogenate of virus-infected embryonating eggs such as Infectious bursal disease virus and Avian papillomavirus (Tables 1, 3). However, all the preparations that contained Fowl pox virus produced a distinct smear in the agarose gel without any loss of the analytical sensitivity (Fig. 1F, G). This effect may result from the fact that pox viruses are DNA viruses and, therefore, vaccines would contain a higher level of DNA or from a special additive often added to the poxvirus vaccines. In another context, the detection sensitivity of the PCR assay varied to some extent according to the type of vaccine matrix (Fig. 1).
In conclusion, the current report describes the development of a highly specific, sensitive, and reproducible PCR assay for the detection of CAV contamination in avian virus vaccines. The assay could be routinely used for evaluation of the purity of vaccine batches, particularity those prepared in chicken embryos, with great impact on the time, labor, and economy of the quality control procedures.
Footnotes
a.
Nobilis® CAV P4, Intervet/Schering-Plough Animal Health, Boxmeer, The Netherlands.
b.
DNASTAR Inc., Madison, WI.
c.
Integrated DNA Technologies Inc., Coralville, IA.
d.
Metabion International AG, Martinsried, Germany.
e.
GF-1 blood DNA extraction kit, Vivantis, Selangor DE, Malaysia.
f.
ABgene, Epsom, Surrey, United Kingdom.
g.
Applied Biosystem Inc., Foster City, CA.
h.
IMAGO Compact Imaging System, B&L Systems, Maarssen, The Netherlands.
i.
GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom.