
Abstract
The present study aimed to produce the relatively conserved central fragment of the Mycoplasma gallisepticum PvpA cytadhesin as recombinant antigen and to determine its species-specific diagnostic potential in comparison with the full-length recombinant rPvpA336 protein. For this purpose, a recombinant protein (rPvpA134) consisting of 134 amino acids with apparent molecular mass of 27 kD was produced and highly purified. The rPvpA134 protein was composed of the amino acid residues at positions 133–265 with respect to the wild-type PvpA. Two bi-antigenic diagnostic models based on Western blot and enzymatic rapid immunofiltration assay (ERIFA) were developed to compare simultaneously the diagnostic potential of the recombinant antigens rPvpA134 and rPvpA336. Although 40% of the confirmed rPvpA336-positive chicken sera were detected as reactive with rPvpA134, this protein would be a useful secondary diagnostic antigen with which to confirm species-specific antibody response for monitoring M. gallisepticum infections. It can be concluded from the present study that 2 bi-antigenic models were successfully adapted to the specific diagnosis of chicken M. gallisepticum. Furthermore, by virtue of its simplicity and rapidity, the ERIFA model has multi-antigenic application potential, making it an alternative field test that is widely applicable in the veterinary diagnostic field.
Introduction
Mycoplasmas constitute the smallest self-replicating and “ideal parasitic” microorganisms causing chronic and generally mild infections in human beings and animals. Mycoplasma gallisepticum is a member of the most important avian mycoplasmas, causing chronic respiratory disease in chickens and infectious sinusitis in turkeys, leading to important economic losses in the poultry industry through the reduction of meat and egg production. 12,21 The disease may be accompanied by secondary infections that greatly affect its severity. Periodic surveillance of the infection using diagnostic tests is essential for monitoring and maintaining mycoplasma-free flocks. Diagnostic techniques for determining M. gallisepticum–infected flocks are essentially based on isolation and identification of the bacteria by culture, use of specific DNA detection methods, and demonstration of the presence of specific anti–M. gallisepticum antibody by serological tests. 21
Although culture- and DNA-based detections are considered as confirmatory tests, they are technically challenging, time consuming, and necessitate appropriate infrastructure and skilled personnel. In addition, these techniques are difficult to carry out in the screening of large numbers of samples. Because of their large-scale applicability, serological tests such as rapid serum agglutination (RSA), hemagglutination inhibition, and enzyme-linked immunosorbent assay (ELISA) are most commonly used and recommended for monitoring flocks. 21 However, lack of specificity and/or sensitivity of serological tests often occurs because of antigenic differences between the mycoplasma diagnostic strains and the natural or experimental infecting strains. 5,10 In addition, the cross-reactions also complicate differential diagnosis of mycoplasma infections due to M. gallisepticum and Mycoplasma synoviae, 2 major etiologic agents that share common antigenic determinants. 2,4,15 To overcome these disadvantages, production and use of recombinant proteins of the immunogenic molecules of several mycoplasma strains (or their functional fragments) for the standardization of diagnostic antigen and development of diagnostic tools have been undertaken by several research teams. 3,8,16,17 Recently, the M. gallisepticum PvpA cytadhesin described as surface-exposed immunogenic protein 6,11,23 has been produced in Escherichia coli and used under purified form as a species-specific recombinant antigen (rPvpA336) to develop an individual rapid test for screening M. gallisepticum infections in the field and under limited laboratory conditions. 8
In the present study, a fragment of the pvpA structural gene corresponding to the central region was cloned to produce a recombinant antigen because recent studies 6,8,14,23 have demonstrated the size variability of the PvpA cytadhesin of different M. gallisepticum strains due to major deletions, particularly within the C-terminal and direct repeated regions (DR-1 and DR-2). Immunoreactivity of the recombinant protein consisting of 134 amino acids (rPvpA134) was determined using M. gallisepticum–positive and M. gallisepticum–negative chicken sera. The potential of rPvpA134 as a diagnostic antigen was investigated (in comparison with that of the full-length rPvpA336) by bi-antigenic enzymatic rapid immunofiltration assay (ERIFA) and Western blot (WB) analysis.
Materials and methods
Bacterial strains and expression vector
For cloning of the central region of the pvpA gene, genomic DNA was purified from the M. gallisepticum Pendik strain, a as described earlier. 8 Escherichia coli DH5α and pCold-I bacterial expression vector b were used to produce recombinant protein.
Antibody, chicken sera, and diagnostic tools
Mouse monoclonal anti-HisTag antibody (His-probe [H3]: sc-8036) c was used to detect expression and localization of the recombinant proteins during WB analysis. Chicken sera evaluated as positive or negative with RSA a , d and ELISA e , f were obtained from a private diagnostic laboratory. g The chicken sera were reevaluated using commercial ELISA kits, e according to the manufacturers' instructions for detecting M. synoviae and M. gallisepticum antibodies.
Polymerase chain reaction amplification and cloning of the pvpA134 fragment, DNA sequencing, and nucleotide and amino acid sequence data
Genomic DNA extracted from M. gallisepticum Pendik strain was purified by genomic DNA purification kit
h
(following the instructions of the manufacturer) and used as a template for amplification of central region of the pvpA gene (pvpA134). Forward primer containing SacI (5′-GTGGCACAAGAA
Expression of the recombinant proteins
Competent E. coli DH5α cells were transformed with the pCold-pvpA134 vector, and ampicillin-resistant transformants were selected in lysogeny broth, as previously described. 8 A single colony of the transformant containing pCold-pvpA134 vector was cultured until it reached 0.3–0.4 absorbance at 600 nm. Expression of the recombinant PvpA134 protein was induced by the addition of isopropyl beta-D-thiogalactoside (IPTG) i at a final concentration of 0.5 mM to the culture. The incubation was continued under gentle agitation equivalent to 200 rpm/min in an incubator-shaker at 15°C for 24 hr. Expression of the recombinant PvpA336 protein was also performed using the same protocols. The recombinant bacteria collected by centrifugation at 4,000 × g for 20 min were washed 3 times in 0.15 M NaCl and resuspended in lysis buffer (125 mM Tris–HCl, pH 6.8; 1% sodium dodecyl sulfate [SDS]). Bacteria were boiled for 10 min and then centrifuged at 20,000 × g for 20 min. Supernatants were used to check the expression of recombinant proteins.
SDS–polyacrylamide gel electrophoresis and Western blotting
Crude recombinant E. coli lysates as well as the purified recombinant proteins were separated on 10% polyacrylamide gels and stained with Coomassie brilliant blue or electrotransferred to polyvinyldifluoride membranes j for 1 hr at 0.8 mAmp/cm 2 using a semi-dry transblotter. The membrane strips blocked with 1% gelatin from cold-water fish skin (FG) j in phosphate buffered saline with 0.1% Tween 20 (PBST/FG, pH 7.4) were incubated with either chicken sera at a dilution of 1:200 for 1 hr or anti-HisTag mouse monoclonal antibody c at a dilution of 1:200 for 90 min at room temperature with gentle shaking. The membranes were then incubated with either alkaline phosphatase (AP)–conjugated rabbit anti-chicken immunoglobulin Y (IgY) j or AP-conjugated goat anti-mouse γ chain–specific antibody j solutions for 1 hr at room temperature with shaking. Color reaction was developed with the addition of 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) j for membrane and was stopped by washing with water.
Purification of the recombinant proteins
Crude E. coli lysates expressing recombinant proteins dialyzed against phosphate–Tris–urea (PTU) buffer (0.1 M NaH2PO4, 10 mM Tris, 8 M urea, pH 8) were mixed with 1 ml of Ni2+-nitrilotriacetic acid (Ni-NTA) agarose beads k by gently shaking at 200 rpm at room temperature for 1 hr and loaded into the empty column and washed with PTU buffer (pH 6.3). Two-step elution of the recombinant proteins was performed with PTU buffer at pH 5.9 and pH 4.5. The fractions containing the recombinant proteins were determined by SDS-PAGE. The protein bands corresponding to the rPvpA134 and rPvpA336 proteins were excised from the polyacrylamide gel and incubated in the elution buffer (50 mM Tris HCl, 150 mM NaCl, 0.1 mM ethylenediamine tetraacetic acid, pH 7.5) with shaking. Diffusion into the elution buffer and purity of the recombinant proteins were checked by SDS-PAGE analysis.
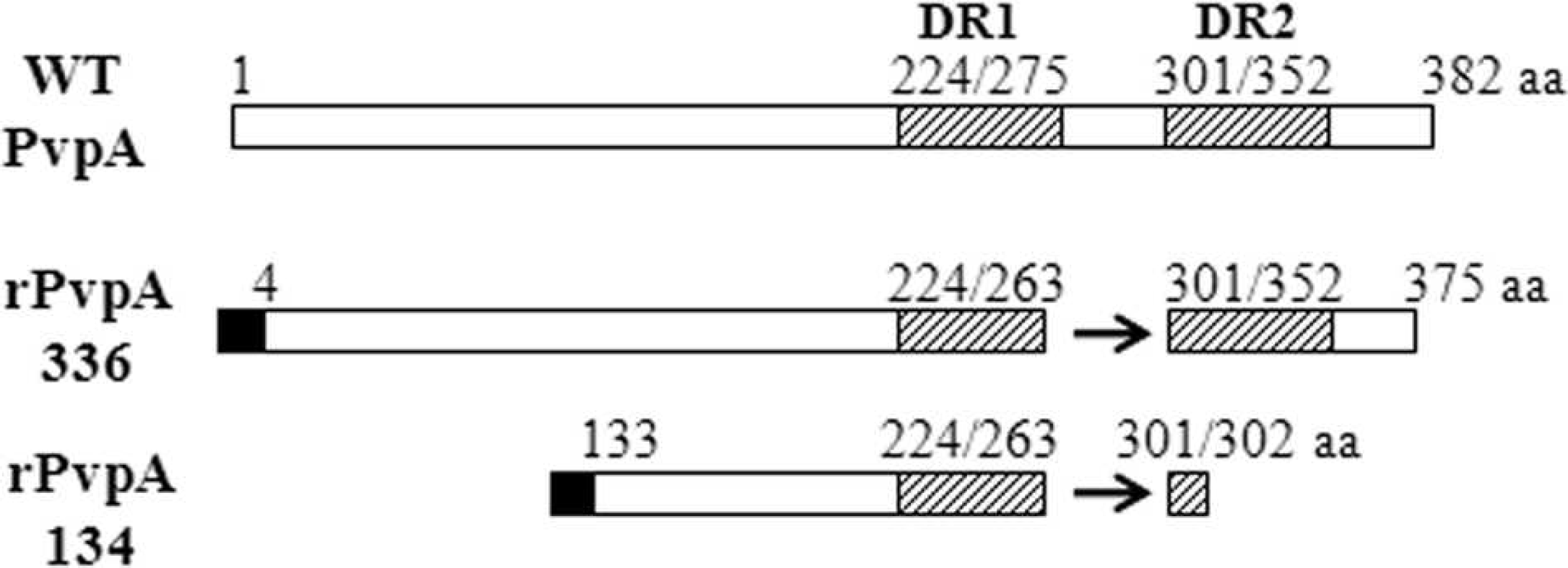
Schematic diagram of the recombinant rPvpA134 protein compared to the wild-type PvpA and the full-length recombinant rPvpA336 proteins. WT = wild-type PvpA. The arrows indicate the deletion region. The numbers show amino acid (aa) positions. Hatched boxes correspond to direct repeat (DR) regions DR-1 and DR-2. Black boxes represent the HisTag part from the pColdI vector.
Enzymatic rapid immunofiltration assay
Enzymatic rapid immunofiltration assay used to detect chicken anti–M. gallisepticum PvpA antibody was performed on a nitrocellulose (NC) membranel included in an individual plastic cassette, as previously described. 8 Briefly, 0.5 μl (1 μg/μl) of highly purified recombinant proteins were separately adsorbed onto the NC membrane as target antigens. After wetting and saturation of the NC membrane with 100 μl of PBST/FG, 50 μl of serum sample was added and flowed through the membrane. Following washing with PBST/FG, 50 μl of AP-conjugated anti-chicken IgY solution j was added and flowed through the NC membrane. The membrane was washed 4 times, and a volume of 50 μl of BCIP/NBT substrate solution j was added. The color reaction developed in 2 min was stopped by washing, and the results were estimated following reaction intensity and visualization time and photographed.
Results
DNA and amino acid sequences of the pvpA134 gene
The PCR-amplified pvpA134 gene fragment was integrated into pCold-I expression vector and then transformed to E. coli cells. The recombinant vector was used as a template for DNA sequence analysis, showing that the pvpA134 gene fragment composed of 402 nucleotides in-frame encodes 134 amino acids localized at the central region of M. gallisepticum PvpA cytadhesin. Figure 1 presents a schematic comparison between the wild-type PvpA (M. gallisepticum R strain, GenBank accession no. AAF67108) and the recombinant proteins of rPvpA134 and rPvpA336. The recombinant PvpA proteins produced from the M. gallisepticum Pendik strain contain an insertion of 1 amino acid at position 165 and a deletion of 37 amino acids at positions 264–300 with respect to the wild-type PvpA. This deletion concerns the last 11 amino acids of wild-type DR region (DR-1, 224–275) and follows 26 amino acid residues until the beginning of the DR-2 region (301–352 amino acid residues). As seen in Figure 1, the rPvpA134 was composed of the amino acid residues at positions 133–263 and 2 additional ones from the position of 301–302 with respect to the wild-type PvpA. Since the residues 301 and 302 were the same at the positions 264 and 265, the rPvpA134 can be considered to contain only DR-1 of the 2 DR sequences. High homology was found between the wild-type and the recombinant PvpA proteins, whereas 9 and 15 amino acid residues in the rPvpA134 and rPvpA336 differ from those of the wild-type PvpA, respectively.
Expression, purification, and reactivity of the rPvpA134 protein
Once DNA sequence analysis showed in-frame cloning of the rPvpA134 gene into pCold-I vector, the transformed E. coli cells were induced by IPTG for recombinant protein production. Sodium dodecyl sulfate–PAGE analysis of E. coli whole-cell extract showed high-level expression of the rPvpA134 protein (Fig. 2, lane 1). Its apparent molecular mass was estimated to be 27 kD compared to the molecular weight marker (Fig. 2, lane M). The rPvpA134 protein was first purified by Ni-NTA affinity chromatography (Fig. 2, lane 2). A further purification was performed to remove slight contaminants of E. coli by elution of the rPvpA134 from the polyacrylamide gel. In this way, the rPvpA134 protein has been highly purified (Fig. 2, lane 3).
The rPvpA134 protein was analyzed by WB using monoclonal anti-HisTag antibody as well as 20 positive and 35 negative chicken sera with the recombinant rPvpA336. Among 20 positive sera, 8 samples were found to be reactive with the rPvpA134 protein, and the negative ones were not reactive. Representative results given in Figure 3 showed the recognition of the rPvpA134 by anti-HisTag antibody (lane HAb) and rPvpA336-positive individual chicken sera (lanes 1–3). In contrast, the rPvpA336-positive samples (lanes 4–5) were detected to be rPvpA134 negative. Lane 6 was an individual negative serum with all tests.
Development of comparative bi-antigenic immunoassays
Two comparative bi-antigenic (rPvpA134 and rPvpA336) immunoassay models, WB and ERIFA, have been developed for simultaneous evaluation of the antigenicity of the purified recombinant PvpA proteins. Enzymatic rapid immunofiltration assay and WB techniques were used as rapid screening and confirmatory tests, respectively. Figure 4 shows the representative results obtained with WB (Fig. 4A) and ERIFA (Fig. 4B) techniques. Using WB analysis, strip HAb blotted with monoclonal anti-HisTag antibody clearly demonstrates the reactivity and presence of a detectable quantity of the recombinant PvpA proteins transferred to NC membrane. Using ERIFA, monoclonal anti-HisTag antibody recognizes the rPvpA134 and rPvpA336 proteins in the same manner (Fig. 4B, cassette 3). As seen in Figure 4A, only a number, but not all, of the rPvpA336-positive chicken sera were found to be reactive with the rPvpA134 protein (lanes 1–4), as demonstrated with individual chicken sera in lanes 5–7. The sample that was not reactive against rPvpA336 was also negative with rPvpA134 (lane 8). The same results were also observed with ERIFA using the sera rPvpA336+/rPvpA134− (cassettes 1 and 7–9) and rPvpA336+/rPvpA134+ (cassettes 2 and 4–6). In testing a total of 20 rPvpA336-positive chicken sera, 8 sera (40%) were found to be reactive with the rPvpA134 protein.
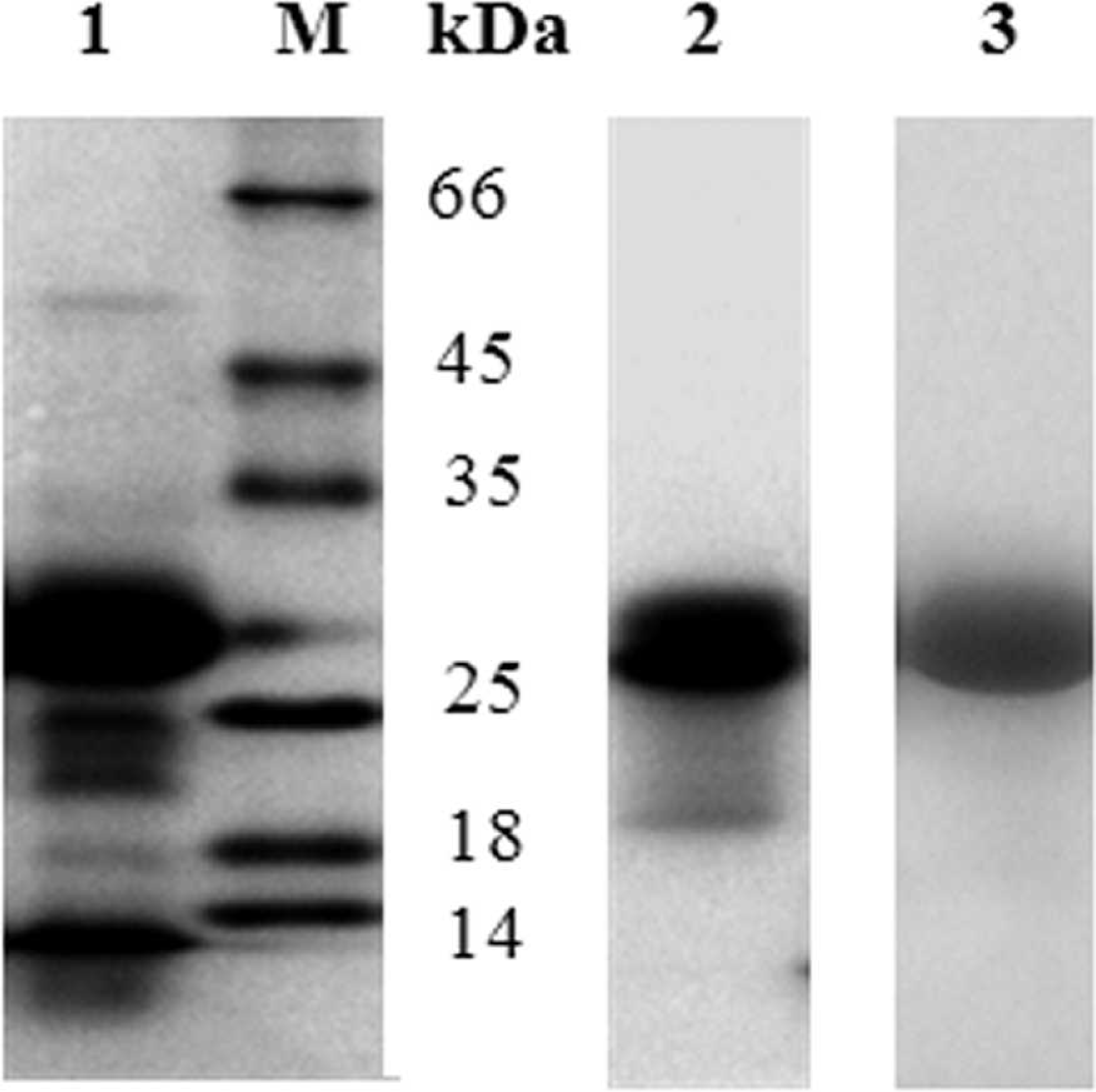
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis of the expression and successive purification of the recombinant rPvpA134 protein by affinity chromatography and gel elution. Lane 1: whole-cell extract of isopropyl beta-D-thiogalactoside–induced bacteria; lanes 2, 3: purified rPvpA134 protein by Ni2+-nitrilotriacetic acid affinity column and polyacrylamide gel elution, respectively; lane M: molecular weight marker.
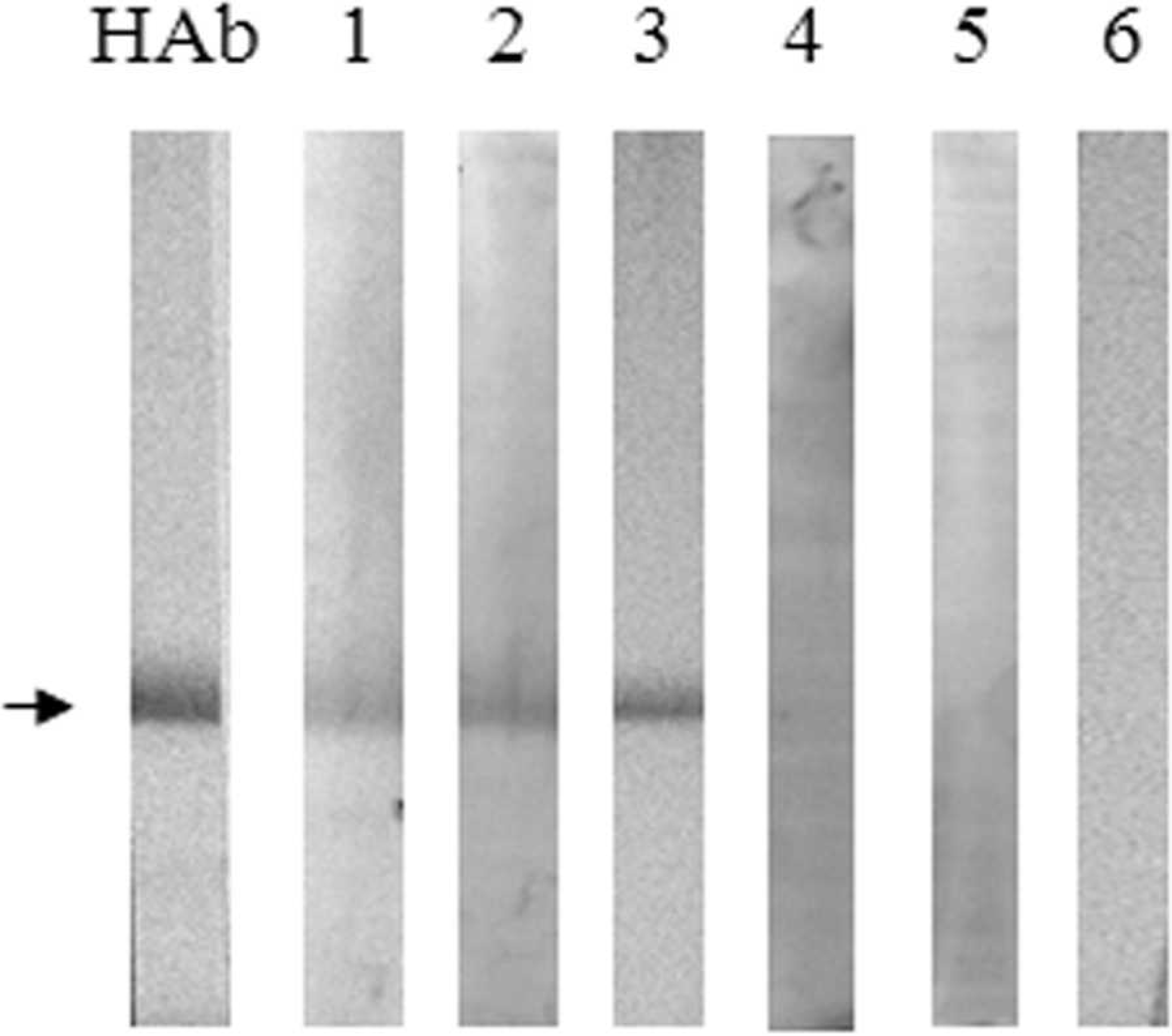
Western blot analysis of the purified recombinant rPvpA134 protein. Lane HAb: monoclonal anti-HisTag antibody; lanes 1–5: rPvpA336-positive chicken sera; lane 6: rPvpA336-negative serum. The arrow shows the rPvpA134 protein.
Discussion
As culture- and DNA-based diagnosis of avian mycoplasmosis necessitates appropriate infrastructure and skilled personnel, diagnosis of M. gallisepticum infections is commonly based on the detection of a specific antibody response. For this reason, several commercial RSA and ELISA kits are used worldwide for monitoring M. gallisepticum infections. However, the accuracy of the serological tests was found to be closely related to the antigenic differences between the mycoplasma diagnostic strains or antigens and the infecting strains. 5,9,10 In addition, the cross-reactions also complicate differential diagnosis of mycoplasma infections due to common antigenic determinants. 2,4,15 Although a heterolog antigen adsorption method has overcome cross-reactions of the polyclonal sera, 2 this method is time consuming and difficult to apply in large-scale seromonitoring. In the last decade, the production of recombinant mycoplasma antigens or their functional fragments allowed the production of standardized diagnostic antigen for the development of accurate diagnostic tools. 3,8,16,17
Although the surface-exposed, species-specific M. gallisepticum PvpA cytadhesin is known as a size-variable protein,6,11,23 its immunogenicity has been demonstrated in native and recombinant forms.6,20,23 Recently, a recombinant PvpA-based (rPvpA336) individual rapid diagnostic tool has been developed for monitoring M. gallisepticum infections in the field and under limited laboratory conditions, and its species-specificity was shown 8 in comparison with chicken sera naturally infected by M. synoviae. Sequence analysis of the cloned pvpA336 gene of the M. gallisepticum Pendik strain (GenBank accession no. DQ989519.2) has demonstrated a deletion of 37 amino acids within the hot-spot deletion domain of the pvpA gene, as previously described 6,14 for various M. gallisepticum strains. In the present study, the central fragment of the PvpA cytadhesin containing 134 amino acids (with apparent molecular mass of 27 kD) was produced (rPvpA134, GenBank accession no. DQ989519.1), highly purified, and tested to determine whether it still preserves antigenicity of the full-length PvpA protein. The choice of the PvpA134 fragment (133–265 amino acid residues) as a potential target antigen was based on the fact that it corresponds to a highly conserved domain (>90% identity), in comparison with several M. gallisepticum strains (e.g., GenBank accession nos. ADC30742.1, AAF67108.1, ACH73018.1, ADC31231.1, and AAS87535.1). In addition, this fragment is less exposed to genetic alterations, such as the deletions observed within the 2 DR regions. 6 In addition, the PvpA134 fragment contains a large number of proline residues (19.4%) and several proline-rich repeat units known to be highly immunoreactive, 1,7,18 and it is accessible to the host immune response because it represents a surface-exposed part of the PvpA cytadhesin.
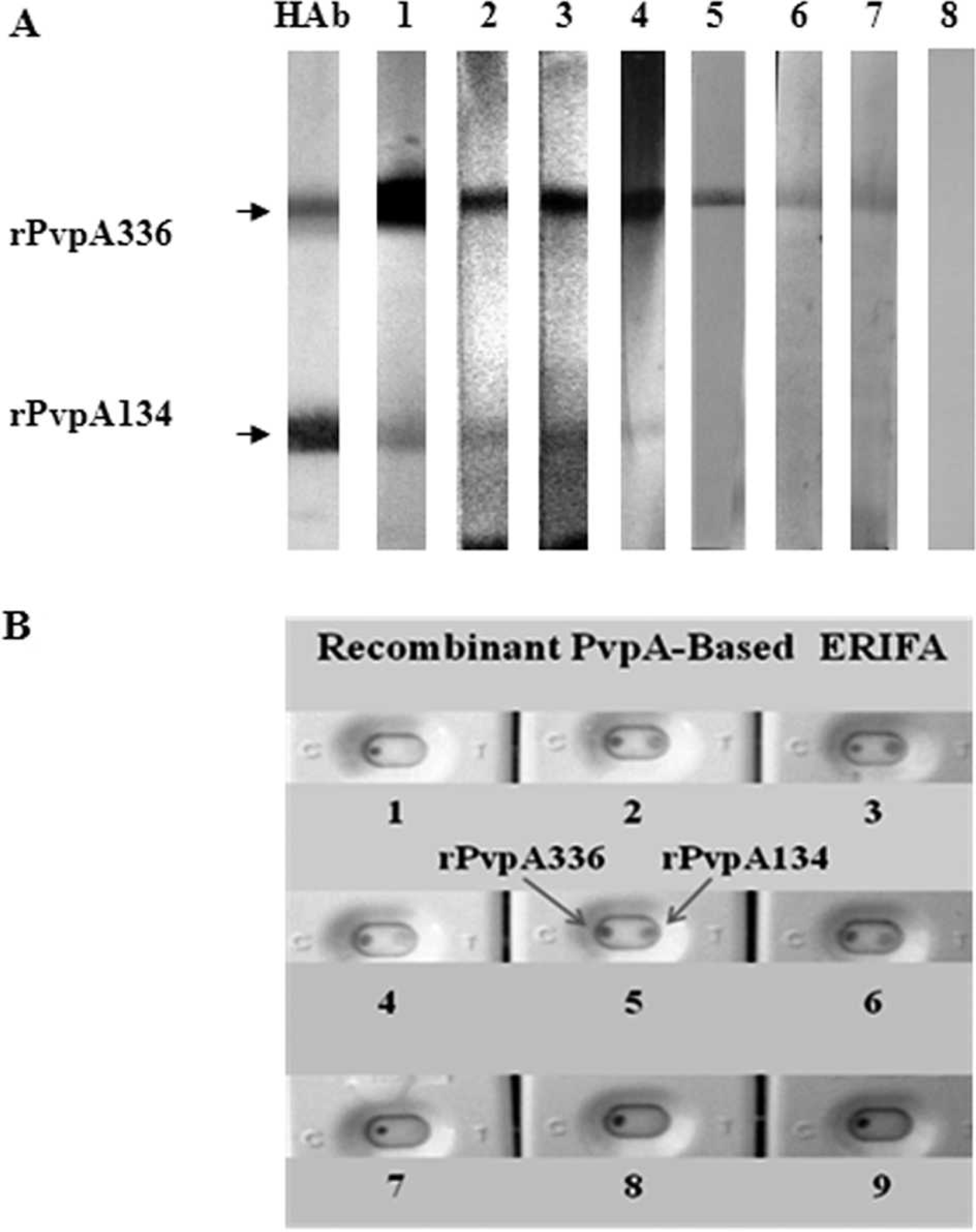
Comparative bi-antigenic immunoassay models for determining immunoreactivity of the recombinant rPvpA134 and rPvpA336 proteins.
In WB analysis with the purified rPvpA134 protein (Fig. 3), monoclonal anti-HisTag antibody and a reduced number (but not all) of the rPvpA336-positive chicken sera were found to be reactive. To confirm these results, indicating that the rPvpA134 partially mimics the antigenicity of the rPvpA336, 2 bi-antigenic diagnostic models were developed for simultaneous evaluation of diagnostic antigens. The models were based on WB and ERIFA techniques by using the rPvpA134 and rPvpA336 as diagnostic antigens. The results with 2 comparative bi-antigenic diagnostic models (Fig. 4) were confirmed by those obtained using mono-antigenic WB analysis. It can be concluded from these results that the central region contains only a part of the antigenic epitopes of the M. gallisepticum PvpA cytadhesin and that full-length PvpA possesses other immunogenic domains located at N- and C-terminal fragments outside the central region. Most likely, the major antigenic domains take place in the C-terminal region containing proline-rich repeat units, known to be highly immunoreactive and involved in the binding of surface-exposed adhesive molecules to host cells. 1,7,13,18 Further investigation is needed to determine the immunodominant fragments of the M. gallisepticum PvpA cytadhesin and to evaluate their adhesive functionality.
While the rPvpA134 mimicking the central region of PvpA cytadhesin was found to be partially immunoreactive in comparison with full-length PvpA, it can be used as a secondary species-specific diagnostic antigen. In this sense, 2 bi-antigenic comparative immunoassays developed in the present study would be useful diagnostic models for monitoring M. gallisepticum antibody response. Although WB is widely used as a confirmatory test, the use of rapid immunofiltration assay is rare in the veterinary diagnostic field. 8,19,22 However, bi-antigenic immunofiltration assay presents a promising individual field diagnostic model by virtue of its simplicity and the rapidity with which it can determine the reactor animals (in less than 5 min in the field and under limited laboratory conditions). In addition, this diagnostic model allows the evaluation of the results by visual inspection. From the results obtained in the current study, it can be concluded that 2 bi-antigenic WB and ERIFA models were successfully adapted to diagnose chicken M. gallisepticum infections. Furthermore, the ERIFA model has multi-antigenic application potential, allowing for the development of alternative screening and confirmatory field tests that are widely applicable in the veterinary diagnostic field.
Acknowledgements. This study was supported by grant 104T271 from the Scientific and Technical Research Council of Turkey (Tübitak, Turkey). The authors thank Dr. C. Yakicier (Bilkent University, Department of Molecular Biology and Genetics, Faculty of Science, Ankara, Turkey) for DNA sequence analysis of the pvpA134 gene fragment.
Footnotes
a.
Pendik Veterinary Control and Research Institute, Istanbul, Turkey.
b.
Takara Inc., Paris, France.
c.
Santa Cruz Biotechnology Inc., Santa Cruz, CA.
d.
Nobilis MG antigen, Intervet-Turkey, Istanbul, Turkey.
e.
BioChek, AN Gouda, Holland.
f.
IDEXX Laboratories Inc., Westbrook, ME.
g.
Protekt Laboratory, Istanbul, Turkey.
h.
Bio Basic Inc., Markham, Ontario, Canada.
i.
Fermentas GmBH, St. Leon-Rot, Germany.
j.
Sigma-Aldrich, St. Louis, MO.
k.
Qiagen GmbH, Hilden, Germany.
l.
Schleicher and Schuell BioScience GmbH, Dassel, Germany.