
Abstract
Collie eye anomaly (CEA) is a canine inherited ocular disease that shows a wide variety of manifestations and severity of clinical lesions. Recently, a CEA-associated mutation was reported, and a DNA test that uses conventional polymerase chain reaction (PCR) has now become available. The objective of the current study was to develop a novel rapid genotyping technique by using SYBR Green–based real-time PCR for future large-scale surveys as a key part in the strategy to eradicate CEA by selective breeding. First, a SYBR Green–based real-time PCR assay for genotyping of CEA was developed and evaluated by using purified DNA samples from normal, carrier, and affected Border Collies in which genotypes had previously been determined by conventional PCR. This real-time PCR assay demonstrated appropriate amplifications in all genotypes, and the results were consistent with those of conventional PCR. Second, the availability of Flinders Technology Associates filter paper (FTA card) as DNA templates for the real-time PCR assay was evaluated by using blood and saliva specimens to determine suitability for CEA screening. DNA-containing solution prepared from a disc of blood- or saliva-spotted FTA cards was available directly as templates for the real-time PCR assay when the volume of solution was 2.5% of the PCR mixture. In conclusion, SYBR Green–based real-time PCR combined with FTA cards is a rapid genotyping technique for CEA that can markedly shorten the overall time required for genotyping as well as simplify the sample preparation. Therefore, this newly developed technique suits large-scale screening in breeding populations of Collie-related breeds.
Keywords
Introduction
Collie eye anomaly (CEA) is a canine inherited ocular disorder characterized by regional hypoplasia of the choroid, the highly vascularized layer of the eye that supplies blood and nutrients to the retina. 14,16,17 The onset of CEA results in an ophthalmoscopically detectable window defect in the ocular fundus located temporal to the optic nerve. Defects of the sclera characterized by colobomatous lesions may also occur and present as pits within or engulfing the optic nerve head or in the adjacent fundus. Mildly to moderately affected individuals appear to retain normal visual function throughout life, as determined by behavioral observation and clinical electroretinography. 7 However, severely affected dogs, particularly those with colobomas, can develop retinal detachments that lead to blindness.
To date, CEA has been described in Collie-related breeds (Border Collie, rough collie, smooth collie, and Shetland Sheepdog) 3,4 and occasionally in other various non-Collie breeds (Australian Shepherd, Beagle, Dachshund, German Shepherd Dog, Lancashire heeler, miniature or toy poodle, and mixed-breed dog). 5,10,12,13 Poor acknowledgment of the clinical significance of CEA is one reason this disease has become so widely spread in many breeds, including collies, and has long been a problem since it was first described in rough collies in 1953. 8 Furthermore, the ambiguous clinical signs and low incidence of blindness related to CEA have led some breeders to erroneously consider that controlling breeding is unnecessary. 4
Collie eye anomaly can be diagnosed by examination of the retina of the eye as early as 5–10 weeks of age. 19 After 12 weeks of age, it is hard to perform an eye examination because the lesion can be covered over with pigment, which makes it impossible to see. In fact, if the proper examination period is missed or there is reason to doubt the accuracy of an ophthalmic examination report, then the condition remains unknown until symptoms become apparent. Moreover, ophthalmoscopic examination can reveal only the affected dogs but not carrier dogs because those conditions are inherited as recessive traits. Therefore, the most definitive diagnostic method for inherited diseases like CEA must be a DNA test. Fortunately, in recent years, the mutation cosegregating with CEA has been identified as a homozygous intronic deletion of 7.8 kilobases (kb) in the NHEJ1 gene, which enables detection of both carriers and homozygotes by a polymerase chain reaction (PCR) based diagnostic test. 11
Primers used in the current study.
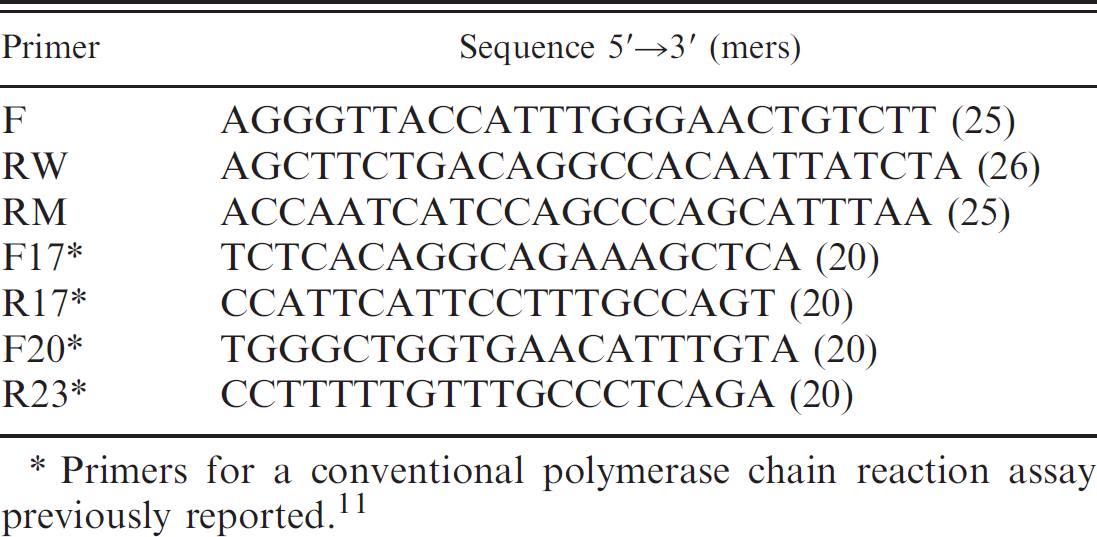
Primers for a conventional polymerase chain reaction assay previously reported. 11
In comparison with conventional PCR assays with gel-based analysis, real-time PCR offers increased sensitivity and specificity in a rapid format. Because of these features, real-time PCR is now one of the most important techniques for detecting and monitoring genetic mutations. Among the different formats available for real-time PCR, the intercalating dye SYBR Green I assay is one of the easiest and most cost-effective methods, because target-specific fluorogenic probes are not required. 21 In addition, simple and rapid sample collection and processing are also indispensable for rapid diagnosis and large-scale screening. Flinders Technology Associates filter paper (FTA card) is a simple technology that reduces the steps required for DNA collection, transportation, purification, and storage, consequently reducing the cost and time required to process a DNA sample to the final step of purified DNA ready for downstream application. 9 The present study developed a SYBR Green–based real-time PCR assay to detect the CEA mutation. This assay was also evaluated for its application to blood and saliva specimens on FTA cards for future large-scale surveys as a measure to prevent CEA.

Schematic diagram of primer locations for genotyping of Collie eye anomaly (CEA) associated deletion. The CEA-associated deletion region is shown within intron 4 on the NHEJ1 gene with flanking and internal primers. F17-R17 and F20-R23 are 2 pairs of primers for a conventional polymerase chain reaction (PCR) assay reported previously. 11 In the SYBR Green–based real-time PCR assay developed in the present study, a shared forward primer (F) and 2 reverse primers (RW and RM) were designed for amplifications of wild-type and mutant alleles, respectively.
Materials and methods
Sample collection
Samples used in the present study were collected from more than 30 Border Collies in several breeding colonies. By using a commercial kit, a DNA samples were purified from crude buccal cell DNA solutions that had been prepared by using another commercial kit b and were stored at −25°C to keep the samples viable for other experiments. The concentrations of the purified DNA samples were adjusted to between 20 and 50 ng/μl. Blood and saliva specimens were also collected from other Border Collies and spotted onto FTA cards for blood c and saliva c specimens, respectively, right after collection. The specimens were dried at room temperature and stored at approximately 4°C until used. Genotype for all the dogs was determined by a conventional 2-step PCR test previously reported. 11
Primer design
As shown in Table 1 and Figure 1, 1 shared forward (F) and 2 reverse primers (RW and RM), placed inside and outside the mutant deletion, were designed for the SYBR Green–based real-time PCR assay. In addition, the other 2 sets of primers (F17-R17 and F20-R23) were used for a conventional PCR previously reported. 11
Conventional PCR protocol
Conventional PCR was carried out in 10 μl of a reaction mixture that contained 5 μl of 2 × Taq polymerase master mixture, d each of the forward and reverse primers at a final concentration of 625 nM, and 1 μl of purified DNA sample. The cycling conditions were an initial denaturing step at 94°C for 5 min, followed by 40 cycles of 94°C for 30 sec, 60°C for 30 sec, and 74°C for 1 min, with a final extension at 74°C for 2 min. The products were electrophoresed on a 2% agarose gel, e along with a DNA ladder, f and visualized by staining with ethidium bromide.
SYBR Green–based real-time PCR
SYBR Green–based real-time PCR with a melting curve analysis was carried out by 2 PCR amplifications for each sample by using a real-time PCR system. g One reaction that uses F and RW primers was designed to yield a 120-bp product that corresponds to wild-type allele, and the other reaction, by using F and RM primers, was designed to yield a 68-bp product that corresponds to the mutant allele. Each reaction was carried out in 10 μl of reaction mixture that contained 2× SYBR Green master mixture, g each of the forward and reverse primers at a final concentration of 625 nM, and 1 μl of purified DNA sample. The thermal profile began with incubation at 95deg;C for 20 sec (predenaturation) followed by 50 cycles of amplification alternating between 95°C for 3 sec (denaturation) and 70°C for 30 sec (annealing/extension). The SYBR Green fluorescent signal was obtained once per cycle at the end of the extension step. After amplification, melting curve analysis was performed by heating the PCR products to 95°C for 15 sec, then cooling it to 60°C for 1 min, followed by a linear temperature increase to 95°C at a rate of 0.3°C/sec while continuously monitoring the fluorescent signal. Data were analyzed by the standard software g included with the real-time PCR system.
Microchip electrophoresis
After the completion of real-time PCR, the amplified products were analyzed by microchip electrophoresis to confirm the accuracy of the allele-specific PCR amplifications. The experiment was carried out by using a microchip electrophoresis system h with its special reagent kit, h including internal DNA size markers and DNA separation buffer. A fluorescent dye i was added to the DNA separation buffer according to the manufacturer's protocol of the microchip electrophoresis system. DNA ladder f (25–450 bp) was used as a reference for DNA sizing.
Application of FTA cards
Application and limitation of specimens on FTA cards as DNA templates to the SYBR Green–based real-time PCR assay were evaluated in the present study. A disc that measured 1.2-mm in diameter was punched from a blood- or saliva-spotted FTA card by using a hole punch. c DNA-containing solution was prepared from an FTA disc by using a DNA extraction kit g that consisted of lysis and DNA stabilizing solutions. The disc was placed into a separate 0.5-ml tube, lysed in the tube with 8 μl of lysis solution, and subsequently incubated at 95°C for 3 min; 8 μl of DNA stabilizing solution was then added to the tube. This DNA-containing solution was transferred to a new tube and stored at −25°C until analysis.
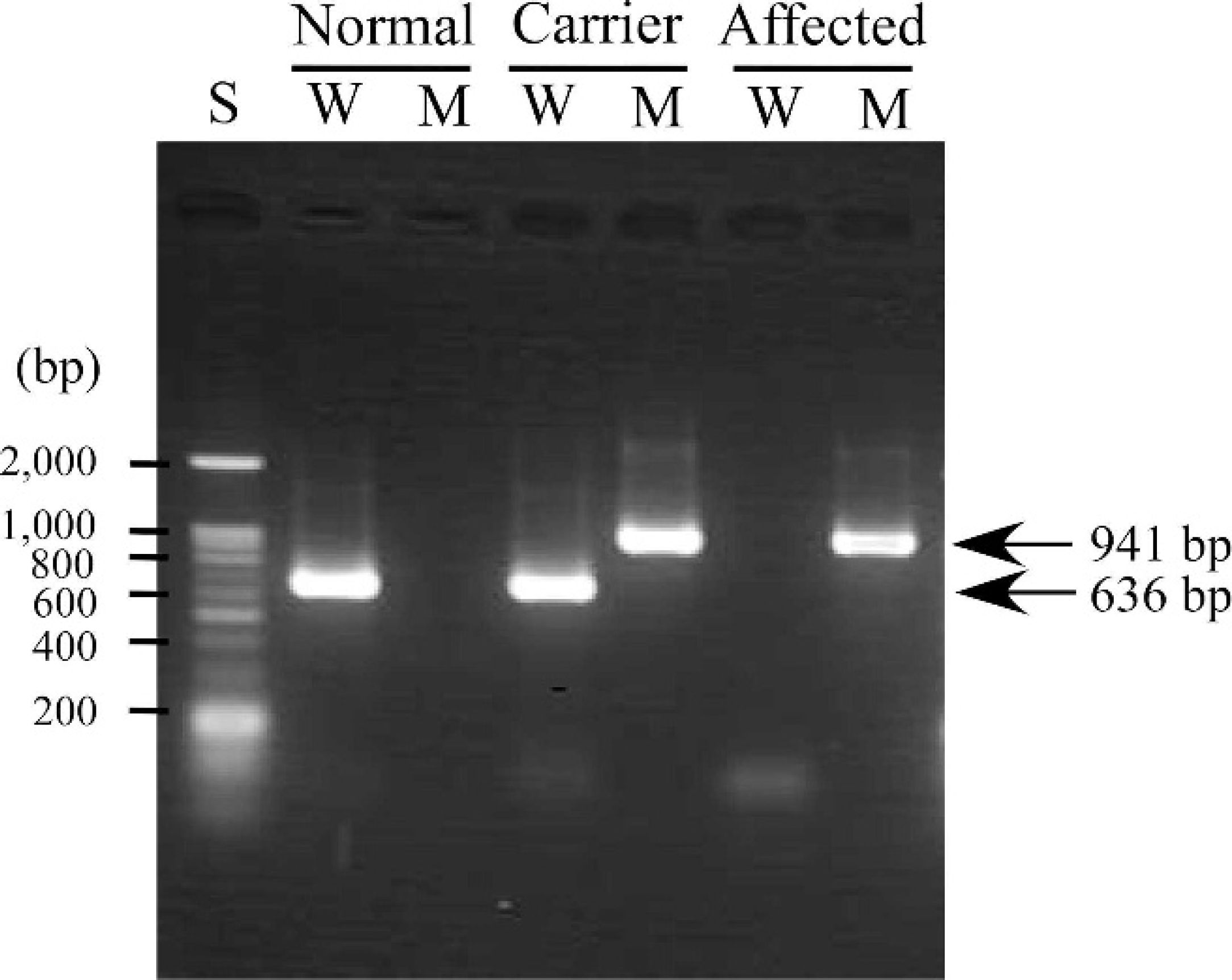
Electrophoretogram with a conventional polymerase chain reaction (PCR) assay. Fragment patterns in normal, carrier, and affected genotypes are shown with size markers (S). The PCR products of W lanes show the results of wild-type allele amplification, whereas PCR products of M lanes show the results of mutant allele amplification.
As a DNA template, 0.25, 0.5, 1, or 2 μl of DNA-containing solution was added to the PCR reaction mixture in a final volume of 10 μl. To evaluate the effect of washing FTA discs before the preparation of DNA, a disc of blood-or saliva-spotted FTA card was washed 3 times with 100 μl of a special reagent j and then twice with 200 μl of Tris–ethylenediamine tetra-acetic acid (EDTA) buffer (pH 8.0). In addition, to evaluate the effect of lysis and DNA stabilizing solutions (extraction mixture) on real-time PCR amplification, this 1:1 extraction mixture was directly added to the PCR reaction mixture with 1 μl of purified DNA sample that was extracted from canine buccal cells, as described above.
Results
Conventional PCR and genotyping of samples
On conventional PCR when using the 2 sets of primer pairs (Table 1, Fig. 1) reported previously, 11 the predicted 636-bp and 941-bp products were obtained that corresponded to wild-type and mutant alleles, respectively (Fig. 2). By using this method, all samples used in the present study could be divided into 3 genotypes (i.e., normal, carrier, and affected).
SYBR Green–based real-time PCR
For optimization of SYBR Green–based real-time PCR, primers were designed to amplify much shorter target regions related to the mutation deletion compared with those in the previous conventional PCR (Table 1, Fig. 1). This SYBR Green–based realtime PCR assay was carried out by using 12 purified DNA samples (5 normal, 5 carrier, and 2 affected) and representative amplification plots of the 3 genotypes shown in Fig. 3, in which allele-specific amplifications carried out in 2 separate tubes were presented as an overlapped plot. Genotypes determined by SYBR Green–based real-time PCR were consistent with those determined when using the conventional PCR assay. In the melting curve analysis after PCR amplification, 2 separate melting peaks that corresponded to wild-type and mutant products, respectively, were obtained by using a representative sample from carrier dogs, although the melting temperatures were very close (Fig. 4).
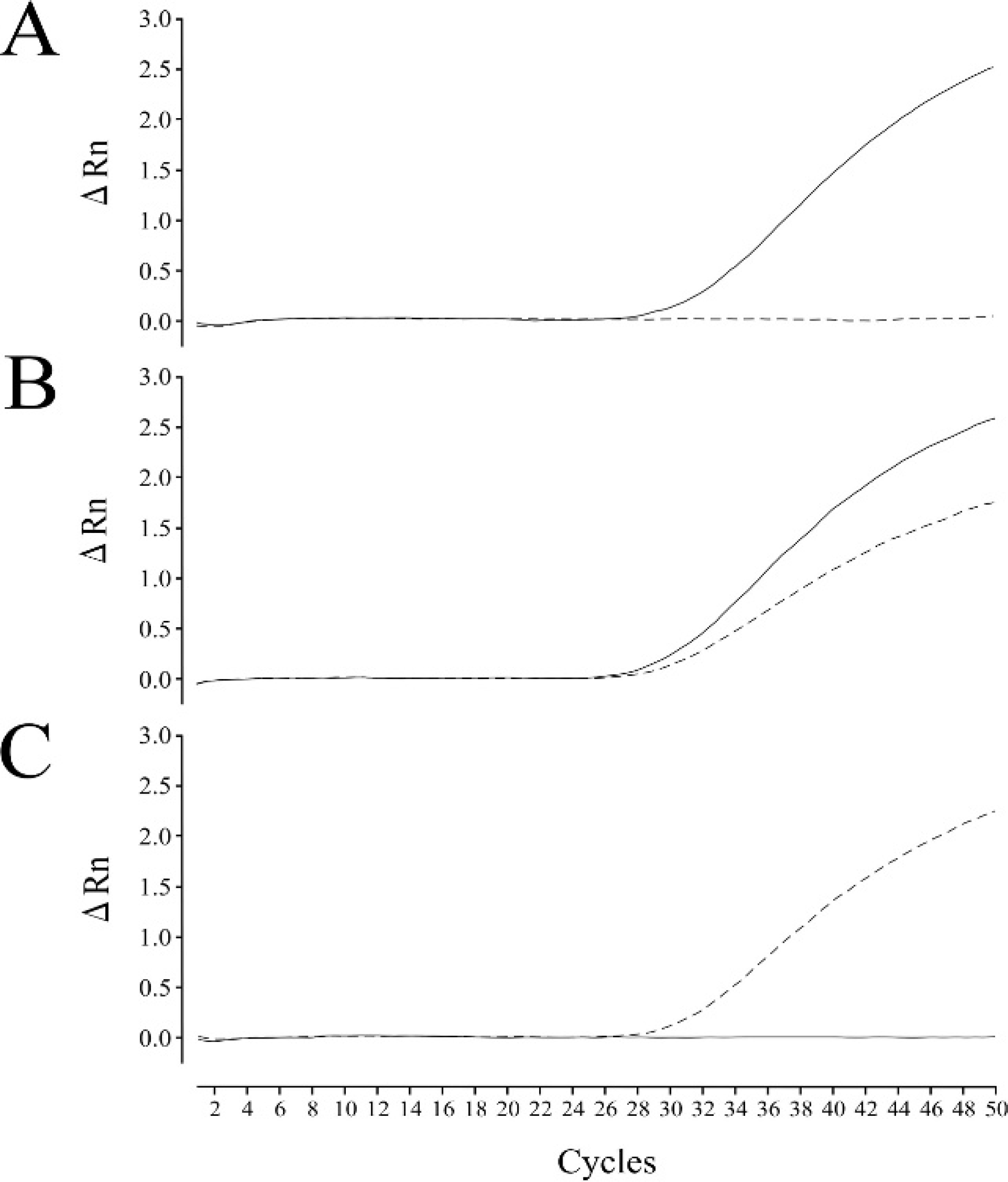
Collie eye anomaly (CEA) genotyping by SYBR Green–based real-time polymerase chain reaction (PCR) assay. Real-time PCR amplifications of wild-type (solid line) and mutant (dotted line) alleles were carried out in 2 separate tubes in the same PCR run and presented in an overlapped plot. Amplification was depicted as fluorescence intensity (ΔRn value) against cycle number. The ΔRn value is the reporter dye signal normalized to the internal reference dye and corrected for the baseline signal established in the first few cycles of PCR. Each of the 3 amplification plots represents normal (
DNA sizing by microchip electrophoresis
he products obtained on real-time PCR were analyzed by using a microchip electrophoresis system h for DNA sizing. Representative electropherographs are shown in Figure 5A, and electrophoretic gel images were reproduced based on the sizing and quantification of DNA fragments (Fig. 5B). As a result, the estimated DNA sizes of the real-time PCR products amplified from wild-type and mutant alleles were the predicted 120-bp and 68-bp fragments, respectively.
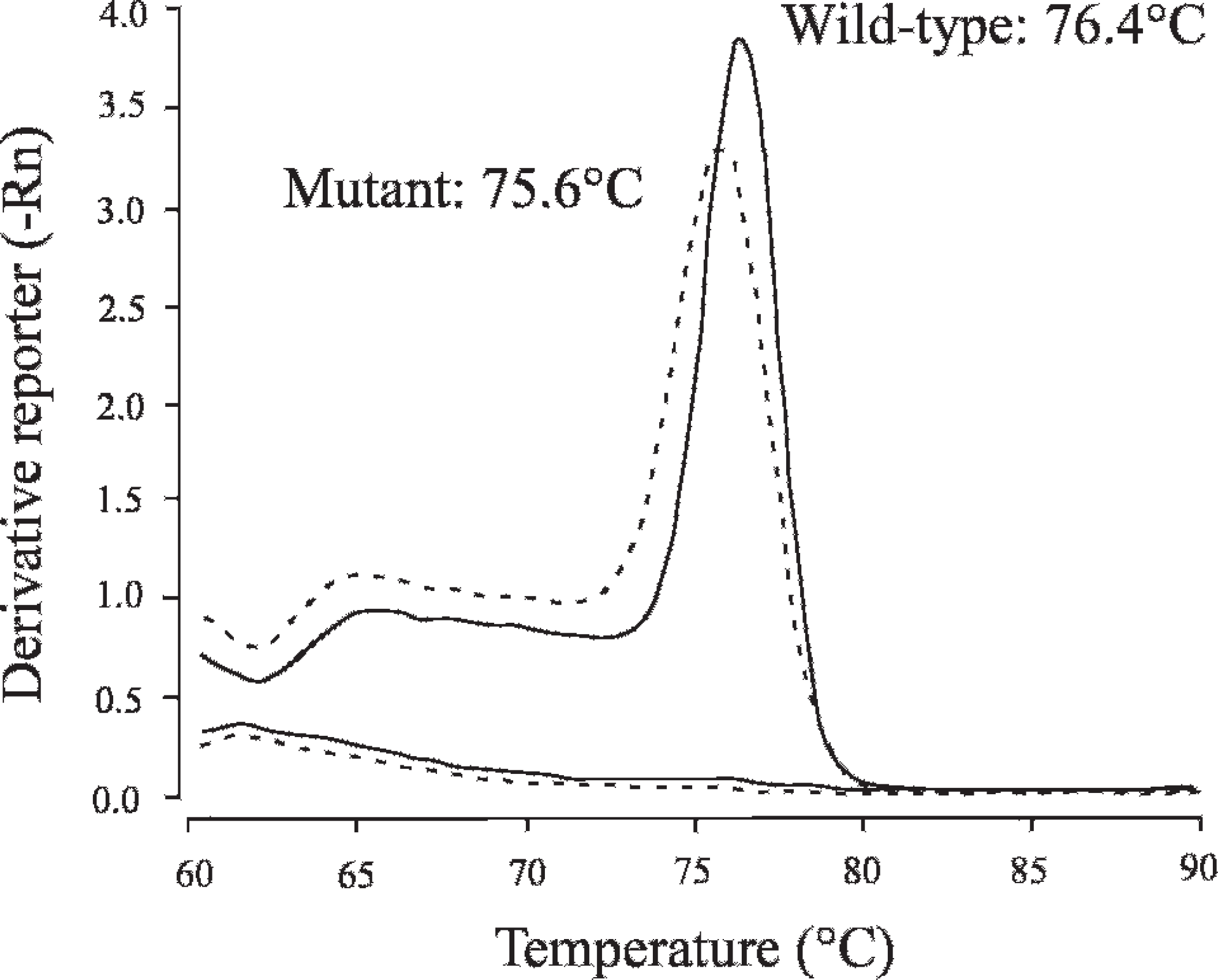
Melting curve analysis in SYBR Green–based real-time polymerase chain reaction (PCR) assay. Solid and dotted lines indicate melting curves of wild-type and mutant alleles, respectively, and representative melting curves of carrier genotype and no template control are presented in an overlapped plot. Two flat curves on the lower side were observed in no template control, which indicated that there were no apparent nonspecific primer-dimers in this PCR.
Application and limitation of FTA cards to SYBR Green–based real-time PCR
When SYBR Green–based real-time PCR was carried out by using 1 μl of DNA-containing solution prepared from an FTA disc as a template in 10 μl of the reaction mixture, weak amplification was observed only in saliva-spotted FTA samples, although there was no apparent amplification in blood-spotted FTA samples (data not shown). Therefore, to evaluate the effects of template solution volume and washing process of FTA discs on PCR amplification, FTA card samples were divided into 2 groups (unwashed and washed FTA) for each of the blood and saliva samples, and 0.25–2 μl of the DNA-containing solution prepared from each FTA disc was added to 10 μl of reaction mixture for real-time PCR. In addition, to evaluate the direct inhibitory effect of extraction mixture (1:1 mixture of lysis and DNA stabilizing solutions), 0.25–2 μl of extraction mixture was also added to 10 μl of reaction mixture with 1 μl of purified DNA sample. The results of these experiments are shown in Figure 6. Amplifications were inhibited as the volume of extraction mixture increased in wild-type and mutant alleles of all samples with or without washing process. Among them, amplifications were severely inhibited when using more than 0.5 μl of DNA-containing solution from unwashed blood-spotted FTA samples, whereas washed blood-spotted FTA samples showed improved amplifications similar to those obtained from saliva-spotted FTA samples. In contrast, the washing process did not make any difference in saliva-spotted FTA samples. When the extraction mixture was directly added to the PCR reaction mixture, amplifications were inhibited as the volume of extraction mixture increased, but the degree of inhibition was intermediate between those of unwashed blood-spotted FTA samples and other FTA samples. With the addition of 1 μl of extraction mixture, amplification was inhibited by 84% and 71% in wild-type and mutant alleles, respectively, compared with those without the addition. When 0.25 μl of DNA-containing solution was used, all samples yielded good amplifications similar to those without the addition of extraction mixture in both wild-type and mutant alleles.
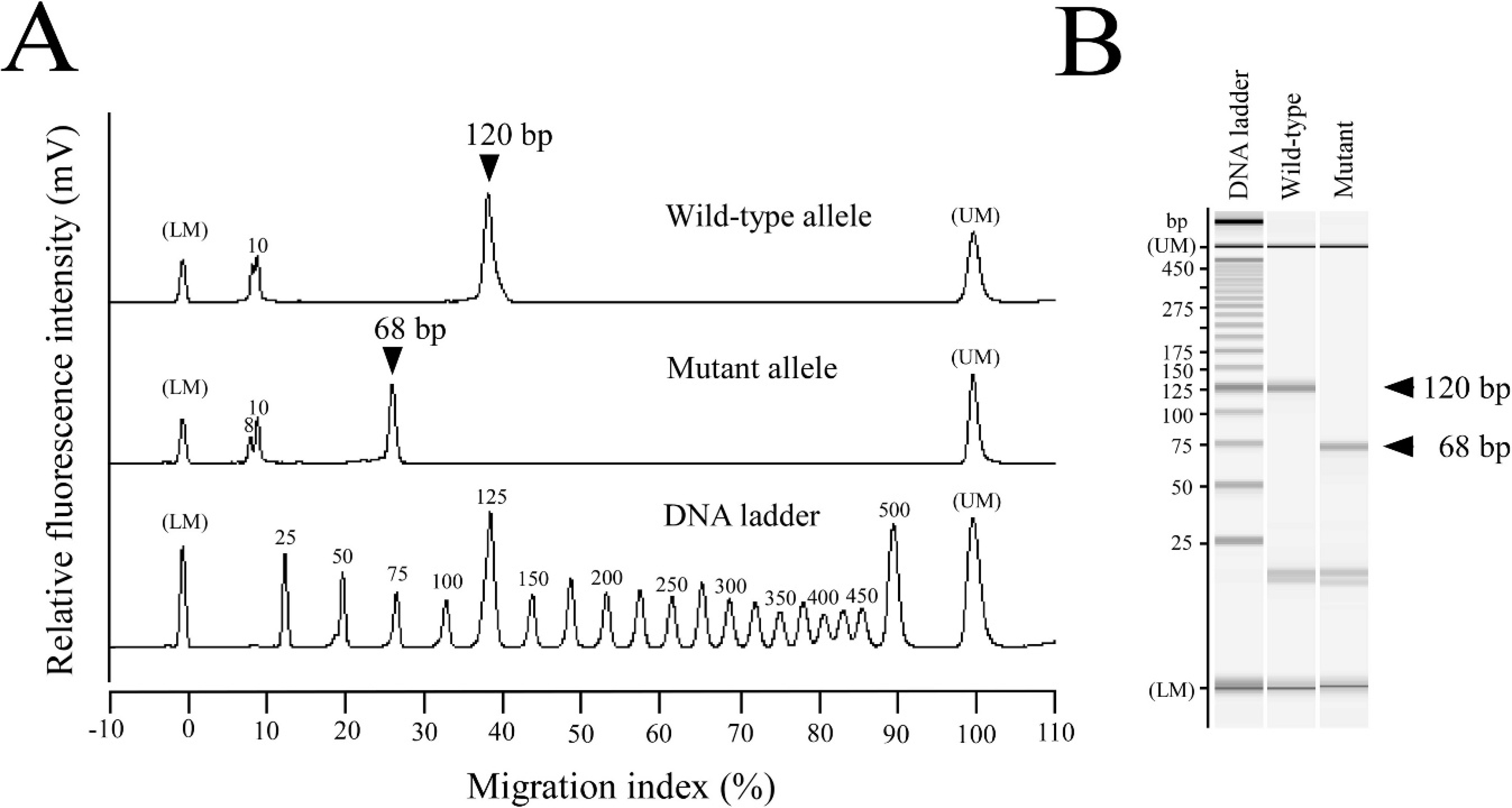
Microchip electrophoresis of the products by SYBR Green–based real-time polymerase chain reaction (PCR) assay.
Discussion
The results demonstrated that the SYBR Green–based real-time PCR assay developed in the current study is available for quick genotyping of CEA. In general, SYBR Green–based real-time PCR assay is less expensive than real-time PCR assay with Taq-Man probes, and less time- and labor-consuming than conventional PCR assay with gel-based analysis. However, there is a disadvantage in SYBR Green–based PCR in that SYBR Green dyes bind to all double-stranded DNA, including nonspecific PCR products and primer-dimers. 21 This characteristic of SYBR Green dyes causes a nonspecific increase in fluorescence signal that is misread as specific amplification, and it is difficult to verify the credibility of the results unlike fragment patterns on agarose gel electrophoresis.
In the present study, melting curve analysis (Fig. 4) and microchip electrophoresis (Fig. 5) were used to verify the credibility of specific amplification in SYBR Green–based real-time PCR assay. The melting curve analysis, carried out automatically after the real-time PCR amplification, determined 2 separate melting temperatures (76.4°C and 75.6°C), which correspond to specific DNA fragments from wild-type and mutant alleles, respectively, and makes it possible to check the specificity of amplifications. However, amplification of nonspecific DNA fragments that have similar melting temperatures may have compromised the authors' judgment concerning the specificity. Microchip electrophoresis can provide more accurate information about the molecular sizes of amplified DNA fragments to verify the specificity of those fragments. Microchip electrophoresis has recently attracted considerable attention for DNA analysis because of its high efficiency, high throughput, time-saving ability, easy operation, and low consumption of samples and reagents. 24 As shown in Figure 5, analysis by microchip electrophoresis clearly exhibited the predicted target sizes of specific DNA fragments that are sufficient to verify the credibility of real-time PCR assay. In addition, the use of microchip electrophoresis does not significantly prolong the total time for genotyping, because automatic analysis can be carried out at a rate of approximately 3 min per sample, unlike agarose gel electrophoresis.
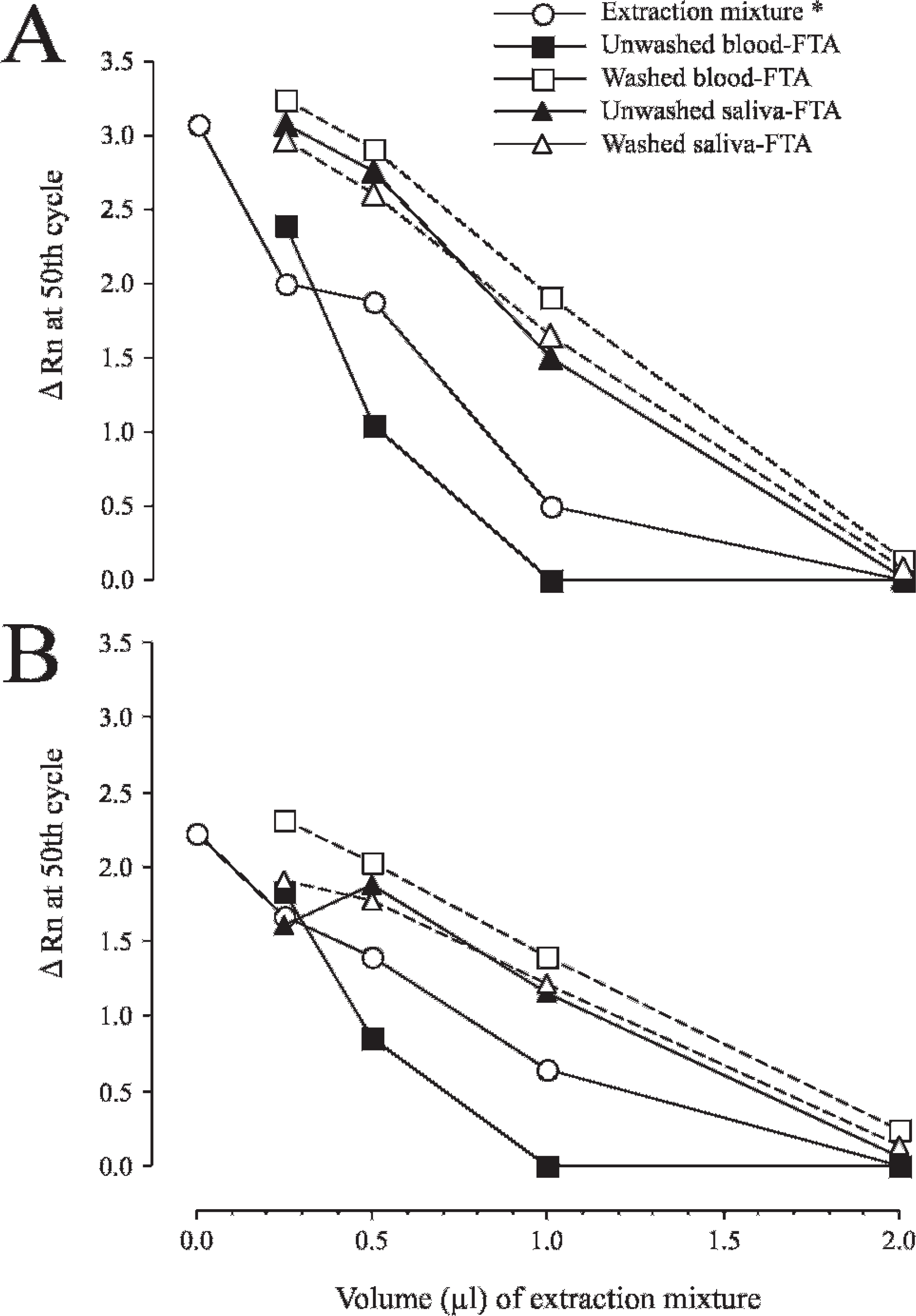
Inhibitory effects of DNA-containing solution prepared from blood and saliva specimens on Flinders Technology Associates filter paper (FTA card) and its DNA extraction mixture on amplifications in SYBR Green–based real-time polymerase chain reaction (PCR) assay. The DNA-containing solution obtained from unwashed or washed FTA card was added to the PCR mixture at various volumes (0.25, 0.5, 1, and 2 μl) as a DNA template. The extraction mixture (*) used to make a DNA-containing solution was directly added to the PCR mixture with 1 μl of purified DNA sample from canine buccal cells as a template. Amplification curves for wild-type (
For assay development, a quick and simple sample preparation procedure is as important as the analytical method to shorten the overall time required for assay. Therefore, application of blood and saliva specimens on FTA cards as DNA templates in SYBR Green–based real-time PCR was evaluated in the present study. As shown in Figure 6, the result in the present study suggested that the extraction mixture, used for extraction of DNA from FTA discs, directly inhibits real-time PCR amplification in a volume-dependent manner. Although there was almost no inhibitory substance in saliva-spotted FTA cards, blood-spotted FTA cards seemed to contain some inhibitory substances, that is, the DNA-containing solution prepared from a blood-spotted FTA disc inhibited PCR amplification most strongly, but washing this FTA disc before DNA preparation dramatically improved the PCR amplification.
In general, biological specimens contain natural inhibitors that interfere with PCR. Blood specimens contain a variety of substances that inhibit PCR, such as heme, 1 immunoglobulin, 2 and anticoagulants, including heparin 6 and EDTA, 18 which might contribute to the inhibitory effect on SYBR Green–based real-time PCR amplification when using an unwashed blood-spotted FTA disc in the present study. Therefore, it is necessary to remove or overcome these inhibitors to allow PCR amplifications to occur efficiently. In the present study, real-time PCR amplification was monitored by using 0.25–2 μl of DNA-containing solution prepared from FTA discs to minimize the inhibitory effect (Fig. 6). When 0.25 μl of DNA-containing solution (2.5% of PCR mixture) was used, sufficient amplification, comparable with that without this solution, was observed. Consequently, it was demonstrated that the washing process of blood- and saliva-spotted FTA discs can be omitted to achieve rapidity and simplicity of the assay, which resulted in the establishment of a rapid genotyping technique for CEA by using SYBR Green–based real-time PCR method applicable to blood and saliva specimens on FTA cards. This enables a future large-scale survey of Collie-related dog populations for CEA by using specimens stored on FTA cards. Furthermore, this method is cost effective: 2.4 and 1.9 times less expensive than the costs of TaqMan probe–based real-time PCR and conventional PCR, respectively.
To date, surveys on CEA or phenotypes of individual dogs have depended mainly on ophthalmoscopic examination and apparent symptoms. 3–5,22,23 According to previous surveys of CEA, the prevalence of affected animals was sufficient to estimate 70–97% for rough and smooth collies in the United States and Great Britain, 4,23 approximately 68% for rough collies in Sweden, 22 72% for Shetland Sheep-dogs, 4 6% for Border Collies, 3 and 13.7% for Lancashire heelers in Great Britain. 5 CEA is a pleomorphic syndrome that shows variability in manifestation, age of onset, and severity of clinical lesions, 15,20 which confuses breeders and ophthalmologists in attempting to determine the phenotype of individual dogs. Moreover, the phenotypes of individuals are not always identical to their genotypes. Practically, there has been some discrepancy between phenotypes and genotypes as shown in Collies in the United States where genotypic incidence was estimated to be as high as 95%, whereas phenotypic incidence has been estimated as 80–85% (Gilger BC: 2006, Diagnosis and treatment of ocular fundus disorders of geriatric dogs. In: Proceedings of the North American Veterinary Conference, pp. 868–870, Orlando, FL).
There have been few published reports about the prevalence of CEA genotypes, although the mutation information of CEA was open to the public. 11 More accurate prevalence based on CEA genotyping results should be clarified in many countries, including Japan. An eradication scheme of CEA should begin with the prevalence survey by using a reliable genotyping assay, and continuous selective breeding based on the survey data is considered as the most effective method of decreasing the high incidences of CEA, leading to the eradication of this disease that is currently widespread in Collie-related breeds.
In conclusion, the present study demonstrated that SYBR Green–based real-time PCR is a rapid and cost-effective genotyping technique for CEA. Moreover, this technique can be available for blood and saliva specimens on FTA cards, which makes it easy to carry out large-scale screening in breeding populations of Collie-related breeds as a measure to prevent the continuing spread of this inherited ocular disease.
Acknowledgements
The authors thank all members of the Japan Border Collie Health Network and for the cooperation of the individual breeders in providing samples. This study was supported financially by grants (nos. 20380173, 20-08112, and 21658109, O.Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) and by the Strategic Research Base Development Program for Private Universities, 2008–2012 (T.A.), which is also funded by MEXT.
Footnotes
a.
DNA Extractor SP Kit, Wako Pure Chemical Industries, Osaka, Japan.
b.
BuccalAmp™ DNA Extraction Kit, EPICENTRE Biotechnologies, Madison, WI.
c.
FTA Classic Card, Indicating FTA Classic Card, Harris Uni-Core Punch (size 1.2 mm); Whatman International Ltd., Piscataway, NJ.
d.
GoTaq® Hot Start Green Master Mix, Promega Corp., Madison, WI.
e.
Agarose for ≥1 kbp fragment, Nacalai Tesque Inc., Kyoto, Japan.
f.
25/100 bp Mixed DNA Ladder, Bioneer, Daejeon, Korea.
g.
StepOne™ Real-Time PCR System, Fast SYBR Green Master Mix, StepOne™ software (version 2.0), DNA Extract All Lysis Reagents Kit; Applied Biosystems, Foster City, CA.
h.
MCE-202 MultiNA Microchip Electrophoresis System, DNA-500 Kit; Shimadzu Corp., Kyoto, Japan.
i.
SYBR Gold Nucleic Acid Gel Stain, 25 bp DNA Ladder; Invitrogen Corp., Carlsbad, CA.
j.
FTA Purification Reagent, Whatman Inc., Florham Park, NJ.