
Abstract
Epizootic hemorrhagic disease virus (EHDV) is a significant pathogen of wild and sometimes domestic ungulates worldwide. Rapid and reliable methods for virus detection and identification play an essential part in the control of epizootic hemorrhagic disease (EHD). In the present study, a 1-step real-time polymerase chain reaction (PCR) group-specific assay was developed. The assay detects genome segment 5 (NS1) from all of the 8 serotypes of EHDV. Assay sensitivity was evaluated relative to a conventional gel-based nested PCR using cell culture–derived virus and diagnostic samples from clinically affected white-tailed deer (Odocoileus virginianus). The assay reliably amplified the NS1 gene from any of the EHDV strains tested, including isolates from each of the 8 EHDV serotypes. No cross-reactions were detected when all 24 serotypes of Bluetongue virus, a closely related member of the genus Orbivirus, were tested. A panel of 76 known EHDV-positive clinical samples was used to compare the performance of the assay relative to a previously reported real-time PCR assay. Results indicated that there was no statistically significant difference between the threshold cycle values obtained with both assays. A collection of 178 diagnostic samples submitted for EHD diagnosis was also used for test evaluation. The assay could be applied for rapid detection of EHDV in clinical samples from susceptible ruminants during an outbreak of the disease. In addition, this PCR assay has the benefits of being reliable and simple and could provide a valuable tool for studying the epidemiology of EHDV infection in susceptible ruminants by facilitating the detection of EHDV, regardless of the serotype.
Epizootic hemorrhagic disease virus (EHDV; family Reoviridae, subfamily Sedoreovirinae, genus Orbivirus) causes a hemorrhagic disease in white-tailed deer (Odocoileus virginianus) and a number of wild ruminants. 2 Although sheep and cattle are susceptible to EHDV, clinical disease has not been reported with circulating North American isolates. The current International Committee on Taxonomy of Viruses lists 8 EHDV serotypes. Molecular studies on EHDV have correlated genetic and serological data from genome segments 2 and 6 that code for the structural proteins VP2 and VP5, respectively. 1,15 These studies indicate that serotype 3 is not a separate group but rather a member of type 1 and, therefore, conclude that there are only 7 distinct serotypes of EHDV. 1 Whereas several serotypes of EHDV can be distinguished worldwide, most cases of hemorrhagic disease observed in white-tailed deer in North America can be attributed to EHDV serotypes 1 and 2. 9
Epizootic hemorrhagic disease virus is transmitted by species of Culicoides midges. This vector dependency has been considered to limit the geographical distribution of the disease. 17 Epizootic hemorrhagic disease virus infections pose a serious threat to wild and captive white-tailed deer populations and cause severe economic losses to the deer industry. 21 Clinical signs of epizootic hemorrhagic disease (EHD) are highly variable; this variation may be associated with viral virulence and host resistance. 13,20 Epizootic hemorrhagic disease outbreaks occur seasonally, but the distribution and severity of these occurrences are highly variable. Morbidity and mortality rates during outbreaks usually are less than 25% but can be 50% or more. The disease characteristically occurs in late summer and early fall, and this seasonality is related to the occurrence of midge vectors. 6 A combination of A combination of case history, clinical signs, lesions, detection of nucleic acid, and the isolation of the virus in suitable host systems are necessary for a diagnosis of EHD. 18 In addition, a rapid test for viral nucleic acid detection can be performed using real-time reverse transcription polymerase chain reaction (real-time RT-PCR) on the original samples submitted. 6
Recently, a new serotype (EHDV-6), previously exotic, has been identified in the United States (Stallknecht DE, Johnson D: 2007, More Interesting HD events from 2006. SCWDS Briefs 23(2): 3–4. Available at //www.uga.edu/scwds/briefs/July07briefs.pdf. Accessed on March 29, 2010). This introduction may be related to the effects of climate change. In Europe, climate change appears to have altered the distribution of Culicoides. 19 There have been no recent studies to determine if the distribution of Culicoides in North America has been affected. The emergence of this new serotype, and the possibility that other serotypes may be present, raise the concern that the geographic distribution of this virus in North America may be changing and that these changes could result in more extensive disease in U.S. wildlife or livestock. 10 Recent reports 24 of disease in Israeli cattle associated with EHDV serotype 7 indicate that different virus strains and/or host factors can affect clinical outcome. The ability to understand the current distribution of serotypes is hindered because small numbers of virus isolations are made from clinical cases as a result of the difficulty in this procedure and the lack of a rapid diagnostic assay for the identification of all existing serotypes of EHDV.
Assays based on RT-PCR have been used to detect EHDV RNA in clinical specimens (e.g., blood or spleen) without the utilization of virus isolation. 2,3,4,11,22,23 These RT-PCR assays are directed toward the highly conserved genes of EHDV of NS1 or NS3 2,5,22,23 or less highly conserved genes such as VP2 or VP3. 3,11 These assays were originally developed for detecting EHDV serotypes 1 and 2 in North America. A real-time PCR group-specific assay, based on the sequence of the gene encoding for the NS3 protein and able to identify all 8 serotypes of EHDV, has been developed. 23 However, this assay uses 7 primer pairs and probes, which can make standardization, optimization, and transfer to other laboratories difficult and time consuming. In an effort to resolve this problem, a simple, rapid, and versatile system is described for the simultaneous recognition and quantitation of all EHDV serotypes directly from clinical specimens.
The EHD viruses used in the current study were grown in baby hamster kidney (BHK)–21 cells and RNA extracted by conventional methods. a The following serotypes and strains were used as reference material to standardize the assay: EHDV-1 (SV123, New Jersey, 3TD); EHDV-2 (ATCC, Alberta, Canada 1962); EHDV-3 (NVSL 047 ODV 0001, Nigeria); EHDV-4 (NVSL 046 ODV 9201, Nigeria); EHDV-5 (NVSL CSIRO 157); EHDV-6 (NVSL C6753); EHDV-7 (NVSL CS775); and EHDV-8 (NVSL DPP059).
Diagnostic specimens (spleen, lung, lymph node) obtained from naturally infected white-tailed deer with clinical signs of the disease were initially homogenized with phosphate buffered saline (PBS; 1:20 w/v) using a bench-top tissue homogenizer b and clarified by centrifugation (600 × g for 5 min). Eighty microliters of the clarified tissue suspension or whole blood were transferred to a 96-well plate. c The RNA was extracted from the samples using a commercially available isolation kit d in a high-speed RNA purification system. c The following 3-step processing program was used, as recommended by the manufacturer d : 1) lysis and binding RNA (containing RNA binding beads) followed by 1 wash with buffer 1 and 2 washes with buffer 2; 2) turbo DNase digestion followed by 2 washes with buffer 2; and 3) elution of RNA with RNase-free water to a final volume of 80 μl.
Primers and TaqMan minor groove binder (MGB) probes for the specific detection of all 8 serotypes of EHDV were designed by multiple sequence aligning of the sequences of segment 5 (NS1) of EHDV serotypes 1–8 (Fig. 1; GenBank accession nos. L27647, X59000, EU928899, EU928900, EU928901, EU928902, EU928903, and EU928904). 14 The primers and TaqMan probes were selected and evaluated using the software Primer Express. d Primers and probes were synthesized, and the stock solution was stored at —20°C until use. d,e The forward (5′-ACWGGVATCATGTTTGAGCT-3′) and reverse (5′-TTCATAACCGCACCTTCATC-3′) primers, corresponding to base positions 1495–1605 of the NS1 gene, were chosen. Probes 1 (6FAM-TCATCACACATCGGC-MGBNFQ) and 2 (6FAM-TCTCGGCATATGCGAGC-MGBNFQ; positions base positions 1516–1550) were designed to detect all 8 serotypes of EHDV (Fig. 1). The PCR amplification was performed by using a 1-step RT-PCR kit. f The mixtures contained 1X buffer, 3 mM MgSO4, 40 pmol forward primer, 20 pmol reverse primer, 4.5 pmol probe 1, 1.5 pmol probe 2, 0.5 μl of Taq mix, and 3 μl of template in a total of 25 μl of reaction volume. Amplification and fluorescence detection were conducted using a sequence detection system d with a program consisting of a reverse transcription step at 45°C for 40 min followed by an inactivation and denaturation step of 95°C for 10 min. The PCR amplification cycle consisted of 45 cycles of 95°C for 15 sec, 50°C for 30 sec, and 72°C for 30 sec, with a final extension step of 72°C for 3 min.
For comparative purposes, a conventional nested PCR was performed 1 using the following primer sequences: P1A: 5′-AAGTTCTTCGTCGACTGCCATC-3′, P1B: 5′-GGTTGCACCATCTGGCCATAAT-3′, P2C: 5′-AGCATTATCACCACAGTGGACG-3′, and P2D: 5′-GCCAATCTATATGCCGCATGT-3′. 2 The PCR conditions were as previously indicated, 22 with several modifications. Briefly, 3 μl of RNA template were used in a 25-μl final volume reaction. The PCR reaction mix contained 1X Taq buffer II (without Mg++), 2.25 mM MgCl2, 2.5 mM of each deoxyribonucleotide triphosphate (dNTP), 20 U of Moloney Murine leukemia virus, 2.5 U of RNasin, i 1 U of DNA polymerase, d and 0.125 μg/μl of each forward and reverse primer. The amplification was carried out in an automated thermal cycler. g The PCR parameters were as follows: 50°C for 50 min, 95°C for 10 min, followed by 30 cycles of 95°C for 30 sec, 60°C for 45 sec, and 74°C for 30 sec, with final extension of 74°C for 7 min. Three microliters of the first PCR products were used for a nested PCR containing 1X Taq buffer II (without Mg++), 2.3 mM MgCl2, 2.25 mM of each dNTP, 5.625 μl of 50% glycerol, 0.15 μg/μl of each nested primer, and 1.25 U of DNA polymerase d in a total volume reaction of 25 μl. The reaction was performed with 1 cycle of 95°C for 10 min, 60°C for 10 sec, and 74°C for 20 sec, followed by 20 cycles of 95°C for 20 sec, 55°C for 10 sec, and 74°C for 20 sec, with a final extension of 74°C for 2 min. The PCR products were loaded on a 2.0% agarose gel. The gel was stained using a fluorescent nucleic acid gel stain. h A DNA band (117 base pairs [bp]) was visualized on ultraviolet transillumination and photographed (Fig. 2).
Analysis of full-length segment 5 of published sequences demonstrated differences between serotypes 1–4 and 5–8. These differences in the NS1 gene facilitated the design of the probes that targeted either serotypes 1–4 or serotypes 5–8 (Fig. 1). Two DNA probes conjugated with a MGB group were designed to amplify a 110-bp fragment of the NS1 gene. The benefit of using these probes is that the MGB group forms extremely stable duplexes with a single-stranded DNA target, allowing shorter probes to be used for hybridization. 25 Because the homology in this gene between serotypes can range between 98% in serotypes 1 and 2 and 77% in the other EHDV serotypes, 23 shorter probes are needed for reliable and efficient detection of all 8 EHDV serotypes. Similar strategies based in the use of short MGB probes 16 or multiple probes 12 to enhance detection of highly variable pathogens have been used for a number of viruses.

Sequence alignment of primers and minor groove binder probes targeted to the NS1 gene of Epizootic hemorrhagic disease virus (EHDV). All sequences are 5′–3′ orientation. Squares indicate areas where forward primers, reverse primers, and probes are bound. Vertical dark lines indicate areas of mismatch within the primers and probes with the template EHDV RNA. W = A/T; V = A/C/G.
The analytical sensitivity of the real-time PCR assay was evaluated using serial viral dilutions. The RNA was extracted and tested as indicated previously. With an arbitrary cutoff threshold cycle (Ct) value of 40, results indicate that the sensitivity of the assay is 0.3–29 fg of the total RNA extracted. EHDV-2, EHDV-3, EHDV-6, and EHDV-8 were detected with the greatest analytical sensitivity (0.3–0.4 fg of RNA). The real-time PCR was less sensitive for EHDV-1 and EHDV-2 (22–29 fg of RNA), with medium sensitivity for EHDV-7 (3 fg of RNA). Because the RNA extraction protocol used in the present experiment does not eliminate all possible RNA and DNA contaminants, the absolute value of the analytical sensitivity may be overestimated. However, the relative sensitivity of the assay between EHDV serotypes can be assessed. The analytical sensitivity of the assay was also evaluated by comparing the relative level of detection with a conventional gel-based nested PCR (Fig. 2). Serial 10-fold dilutions of EHDV were used for RNA extraction, and both conventional and real-time PCR assays were performed. Results indicate that conventional gel-based nested PCR detects only EHDV serotypes 1–4 and 8, in contrast with the real-time PCR, which detected all 8 serotypes of EHDV (Table 1; Fig. 2). The real-time PCR assay is at least as sensitive as the conventional gel-based PCR. However, for EHDV serotypes 2, 3, and 4, the real-time PCR is 1–2 log10 more sensitive than conventional gel-based nested PCR.
The specificity of the assay was evaluated by testing all 24 serotypes of Bluetongue virus (BTV). No amplification was observed with any of the 24 serotypes tested. Twelve white-tailed deer positive to BTV by conventional gel-based PCR were tested. All the samples were negative by EHDV real-time PCR, confirming the specificity of the assay.
In order to evaluate the performance of this assay relative to that of a previously reported real-time PCR for the detection of all 8 EHDV serotypes, 23 an additional panel of 76 clinical samples from cattle and deer naturally infected with EHDV (kindly provided by Dr. W. C. Wilson) were tested and the results analyzed by a Mann–Whitney rank sum test. 8 This nonparametric test was used in order to assess whether the differences between populations means for 2 nonpaired samples were equal. The analysis (Fig. 3) indicates that the difference in the median values between the 2 groups is not great enough to exclude the possibility that the difference is due to random sampling variability. Therefore, there was not a statistical difference (P = 0.103) between these 2 real-time PCR assays, and the null hypothesis that one group does not produce lower Ct values (more sensitive) than the other group cannot be rejected. Threshold cycle values, then, are not likely to be influenced by using either of these 2 real-time PCR assays.
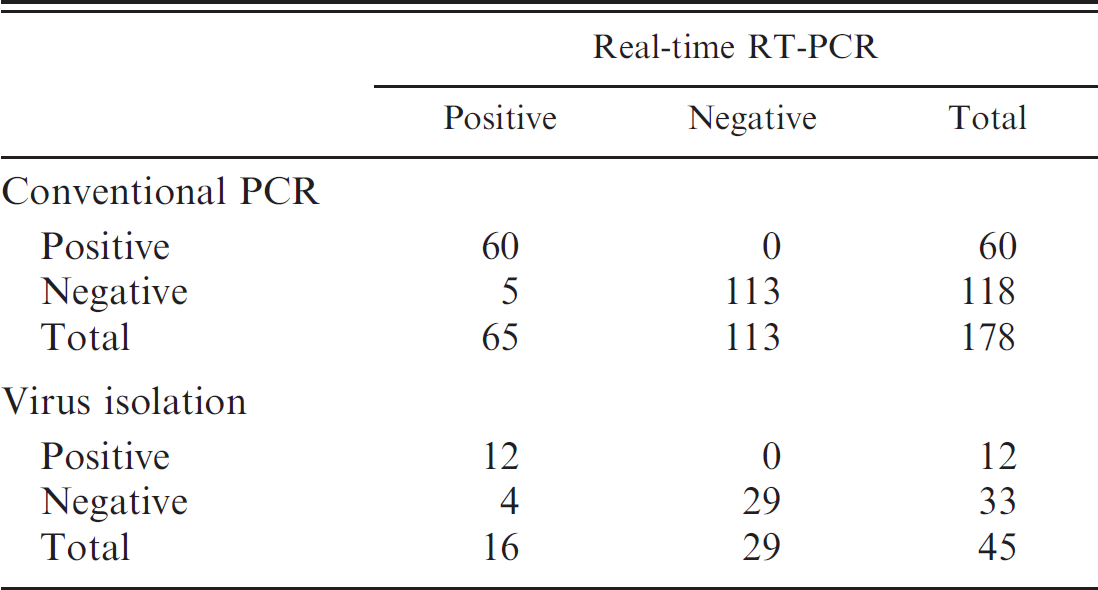
The diagnostic performance of the newly developed assay was evaluated by testing 178 clinical samples from white-tailed deer that have been submitted to the diagnostic laboratory for confirmation of EHD. Samples were tested by both conventional gel-based PCR and real-time PCR as described previously. Using a cutoff value of 40 Ct, the distribution of 60 positive samples in the real-time PCR was between Ct values of 21.5 and 38.7, with a mean of 30.6 Ct. When the 2 assays were compared, the relative diagnostic sensitivity and specificity of the real-time PCR assay were 92.3% (60/65) and 100% (113/113), respectively. The level of agreement was 97.2% (173/178; Table 2). The reduced diagnostic sensitivity of the real-time PCR relative to the conventional gel-based PCR may be related to the high analytical sensitivity of the real-time assay, as The indicated earlier (Table. 1), particularly for EHDV-2, which has been reported as the most prevalent serotype in the southern United States. 6 Forty-seven samples were randomly selected for virus isolation in order to evaluate the performance of the real-time PCR assay relative to a gold standard. Briefly, selected tissues were emulsified using a tissue grinder, with Dulbecco PBS as diluent, to produce a 10% w/v suspension. This suspension was clarified by centrifugation at 2,000 × g at 4°C for 20 min. The samples were then diluted 10–1 and used to inoculate calf pulmonary artery endothelial and BHK-21 cell cultures. Infected cells showed extensive cytopathic effect (CPE) after 3–5 days of culture. The BHK-21 cells showing CPE were confirmed positive for EHDV antigen using the direct fluorescent antibody test. Up to 3 blind passages were done when CPE was not observed before samples were reported negative. Results indicate that there was 91% agreement between the 2 assays. However, as expected, the real-time PCR assay gave 25% (4/16) more positives than did virus isolation, with a sensitivity and specificity relative to virus isolation of 75% (12/16) and 100%, respectively (Table. 2).
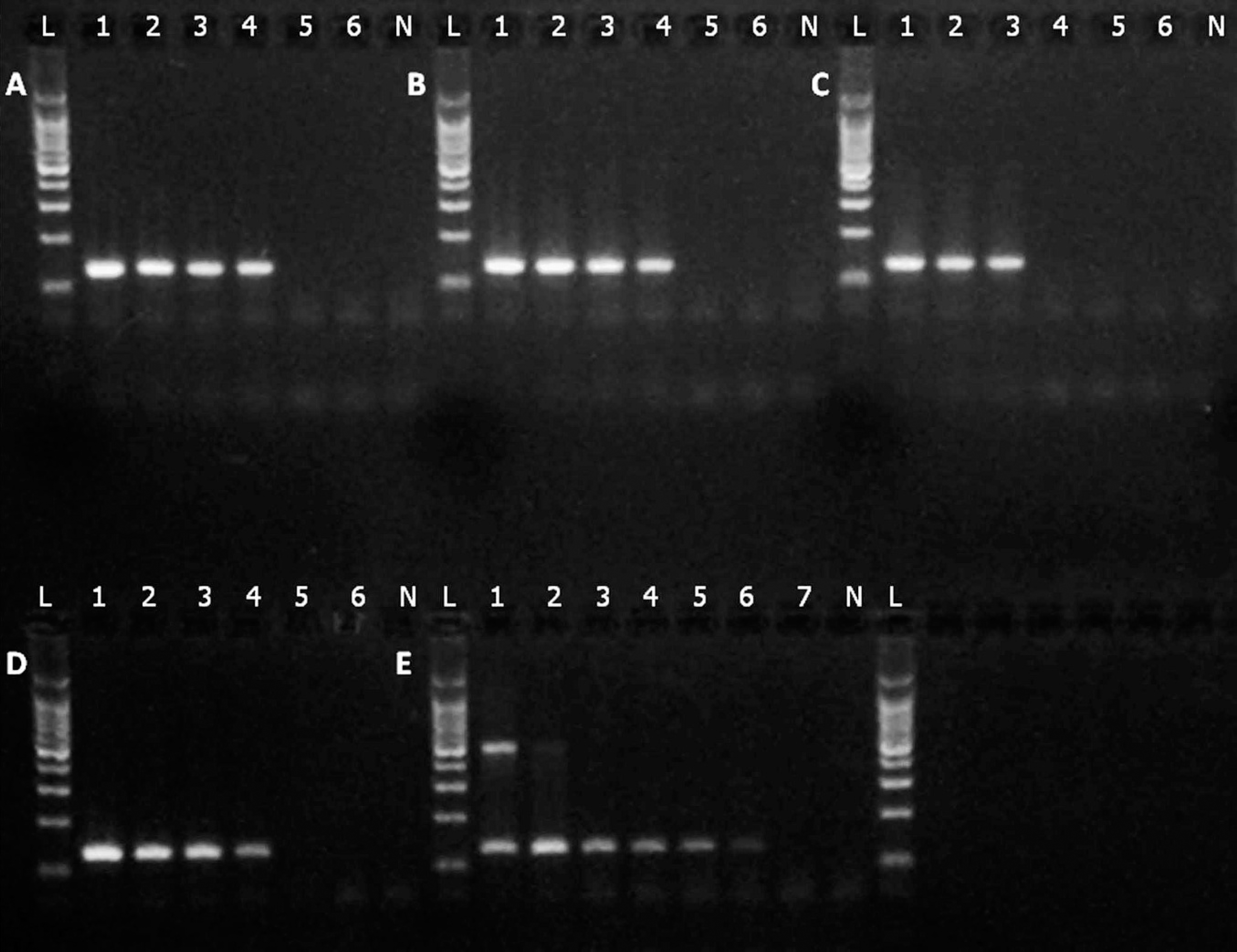
Agarose gel representation of the conventional gel-based polymerase chain reaction (PCR) for serotypes (
Comparative analytical sensitivity of the real-time reverse transcription polymerase chain reaction (RT-PCR) assay relative to the conventional gel-based nested PCR.*
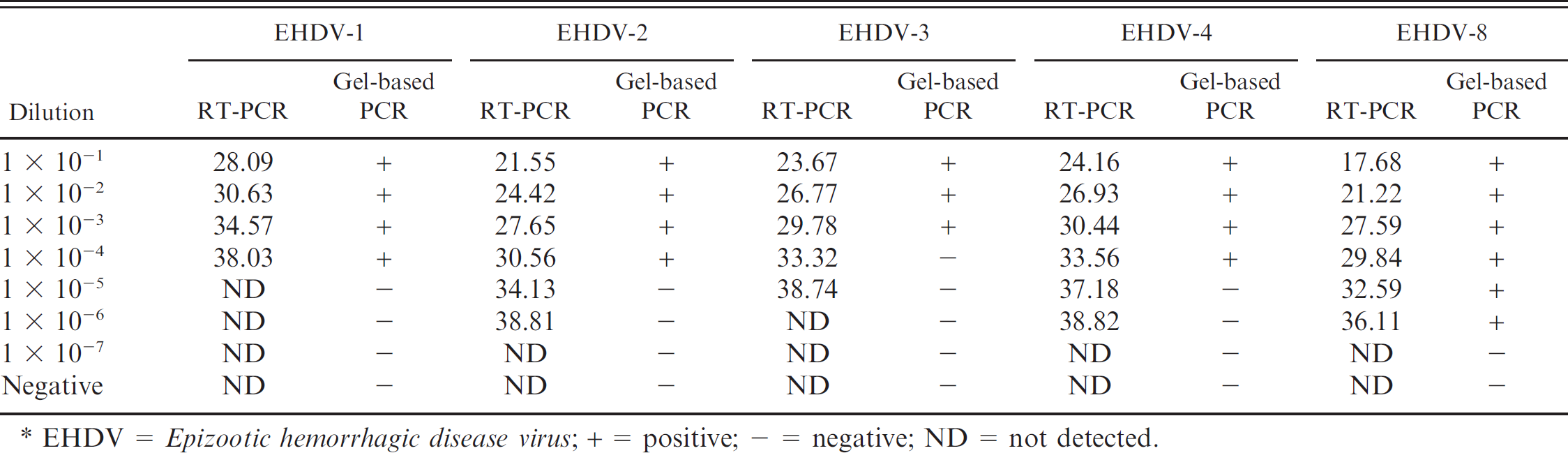
EHDV = Epizootic hemorrhagic disease virus; + = positive; – = negative; ND = not detected.
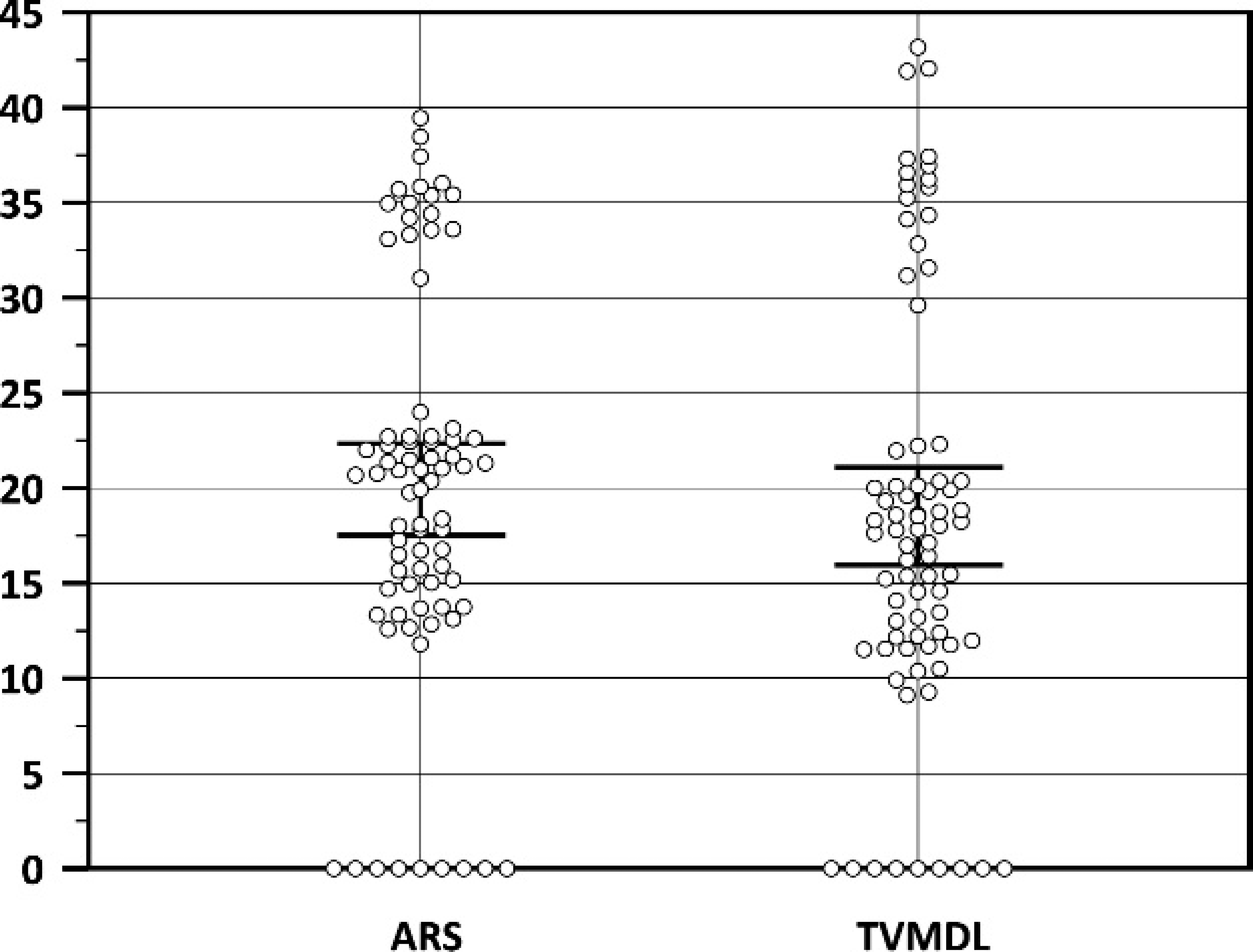
Comparative threshold cycle (Ct) value distribution of 76 clinical samples tested both by a previously reported real-time polymerase chain reaction (PCR) assay 23 and the realtime reverse transcription PCR reported herein. The difference in the median values between the 2 groups is not great enough to exclude the possibility that the difference is due to random sampling variability; there is not a statistically significant difference (P = 0.103). The error bars indicate the 95% confidence interval value of the mean. Not detected is shown as Ct = 0. ARS = U.S. Department of Agriculture, Agricultural Research Service, Arthropod-Borne Animal Diseases Research Unit, Manhattan, KS; TVMDL = Texas Veterinary Medical Diagnostic Laboratory, College Station, TX.
Laboratory diagnosis of EHDV is currently done using a variety of methods. Detecting EHDV in clinical samples is done by virus isolation in cell cultures. Various RT-PCR assays have been developed for detecting EHDV sero-types, particularly those that are endemic in North America. 2,3,4,7,11,22 A molecular method, based on the use of a large number of primer pairs and probes of the gene that encodes for the NS3 protein, with the ability to identify all 8 serotypes of EHDV has been developed. 23 In an effort to increase the simplicity of the protocol and reduce the cost of the assay, a simple, rapid, and versatile real-time PCR assay is described for the simultaneous recognition and quantification of all EHDV serotypes directly from clinical samples. This assay could provide an alternative or a complement to the methods currently used for detection of EHDV infection from a variety of clinical samples. Complemented with gene sequencing, it can be used as a valuable tool to study the epidemiology of EHDV as well as for monitoring emerging serotypes of EHDV in wild animals and domestic livestock. The distribution of EHDV serotypes in North America can be expected to exist in a state of continuous change as climatic changes, especially those relating to temperature, rainfall, and wind patterns, affect the distribution of vectors and hosts. In addition, changes in land use, particularly those related to farmed wild ruminants, have increased considerably in recent years, thereby affecting the geographical distribution of these susceptible species and influencing the areas where new EHDV serotypes can emerge and perhaps persist.
Comparative performance of the Epizootic hemorrhagic disease virus real-time reverse transcription polymerase chain reaction (RT-PCR) assay relative to conventional gel-based nested PCR and virus isolation in cell culture.
Epizootic hemorrhagic disease virus and BTV are 2 of the most important causes of highly infectious noncontagious hemorrhagic disease in wild and captive deer populations in the United States. Although cattle are susceptible to infection, clinical hemorrhagic disease is rare. In addition, infection with either EHDV or BTV can carry restrictions on the sale of livestock and germplasm in international markets. As a result of the simplicity of the newly developed assay in the current study, it may be possible to use this assay simultaneously with a real-time PCR for the detection of BTV in a multiplex format. Such a test could facilitate the simultaneous detection and differentiation of these hemorrhagic infections among susceptible North American deer and livestock populations.
Acknowledgements
The authors thank Janell Kahl, Theresa Ratay, and Shirley Byrne for their excellent technical assistance. Donna Johnson and Dr. Eileen Ostlund from the Animal and Plant Health Inspection Service–National Veterinary Services Laboratory (Ames, IA) are also acknowledged for providing exotic strains of EHDV and RNA from exotic strains of BTV necessary to complete this study. This work was supported by funds from the Texas Veterinary Medical Diagnostic Laboratory and by U.S. Department of Agriculture–Agricultural Research Service project 5430-32000-002. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the U.S. Department of Agriculture.
Footnotes
a.
TRIzol LS Reagent, Invitrogen Corp., Carlsbad, CA.
b.
Seward Stomacher 80 Biomaster Lab Blender, Brinkmann Instruments Inc., Riverview, FL.
c.
KingFisher® 96, Thermo Electron Corp., Waltham, MA.
d.
MagMAX™-96 Blood RNA isolation kit, ABI PRISM® 7000, AmpliTaq-Gold DNA Polymerase; Applied Biosystems Inc., Foster City, CA.
e.
Sigma-Aldrich Corp., St. Louis, MO.
f.
SuperScript III Platinum One-Step qRT-PCR Kit w/ROX, Invitrogen Corp., Carlsbad, CA.
g.
PerkinElmer, Waltham, MA.
h.
GelRed™, Phenix Research Products, Candler, NC.
i.
Promega Corp., Madison, WI.