
Abstract
An indirect enzyme-linked immunosorbent assay (ELISA) by using recombinant Caprine arthritis encephalitis virus (CAEV) p55gag antigen (rELISA), an indirect ELISA by using whole CAEV (wELISA), and Western blot analysis by using the recombinant p55gag antigen (rWB) were developed for detection of CAEV-specific antibodies in goats. Seven hundred and forty-five sera from goats were tested by rELISA, wELISA, rWB, and agar gel immunodiffusion test (AGID), and the results were compared with those of WB analysis by using the whole CAEV antigen (wWB). The AGID test and rWB had similar sensitivities of 93.3% (95% confidence interval [CI]) and 93% (95% CI), respectively, and similar specificities of 96.0% (95% CI) and 96.3% (95% CI), respectively, compared with wWB. The wELISA had substantially lower sensitivity (80.4%) and specificity (78.0%) compared with wWB, and rELISA had the lowest sensitivity (78.2%) and specificity (61.1%) compared with wWB. The lack of adequate sensitivity and specificity for rELISA and wELISA suggests that these assays need considerable modification. However, the results for rWB show that this assay has excellent agreement with wWB and that it can be used as a confirmatory test for the presence of anti-CAEV antibodies.
Caprine arthritis encephalitis virus (CAEV; family Retroviridae, subfamily Orthoretrovirinae, genus Lentivirus) and other viruses in this genus, including Visna/maedi virus of sheep and Equine infectious anemia virus, cause persistent infection and various diseases in mammals. Caprine arthritis encephalitis virus and Visna/maedi virus are genetically distinct but antigenically related; thus, these viruses are designated small ruminant lentiviruses (SRLV). 3,8,14 Caprine arthritis encephalitis virus causes arthritis, interstitial pneumonia, and indurative mastitis in goats, although most infected animals remain symptomless. However, regardless of the symptoms, the virus is transmitted from infected animals to other goats via colostrum or milk, blood, and secretions of the upper respiratory tract. 3 Virus isolation is not efficient, because propagation of SRLV in culture takes time and sometimes the viruses do not show cytopathic effects. 17 Therefore, diagnosis by detecting specific antibodies or viral genes is more efficient than that by isolation of the virus or clinical observation in the control of SRLV infection. 7 The World Organization for Animal Health recommends antibody detection by using serologic tests, such as the agar gel immunodiffusion (AGID) test and enzyme-linked immuno-sorbent assay (ELISA). 23 In Japan, an AGID test has been used for diagnosis of CAEV infection since the first outbreak in 2002. 11 Although commercial ELISA kits for CAEV are available in several countries, 7 these out-of-country kits are unavailable in Japan. Moreover, results of previous studies suggest that the virus strain used in ELISA as the antigen may fail to detect antibodies against a particular strain because of differences of genotypes among the field strains in each country. 1,19 Hence, it is essential to develop ELISA by using the Japanese-originated isolate as the antigen for large-scale screening tests to diagnose CAEV in Japan.
In the current study, an ELISA method was developed by using recombinant p55gag, a major precursor of the CAEV core proteins, as the antigen (rELISA). Furthermore, another ELISA that used the whole viral particle of CAEV isolated in Japan (wELISA) was also developed for comparison with the rELISA.
The precursor of the core protein (p55gag) was chosen as the antigen for the rELISA and the recombinant protein Western blot analysis (rWB). A Japanese isolate, CAEV strain 40 (CAEV40), which was isolated from a naturally infected goat with arthritis, was propagated in fetal lamb lung (FLL) cells 11 and proviral DNA was extracted from the infected cells. The entire genetic region that encoded p55gag was amplified by polymerase chain reaction (PCR) with a primer pair that contained the sequence for either BamHI or SalI. The sequences of the primers used were 5′-GGATCCATGGTGAGTCTAGATAGAG-3′ (forward primer) and 5′-GTCGACTTATTCCATAGGAGGAGCG-3′ (reverse primer). A commercial Taq DNA polymerase kit a was used for the PCR reaction mixture, and amplification was done in 50 μl of a mixture that contained 5 μlof 10 x Taq buffer (including 20 mM Mg2+), 0.2 mM deoxyribonucleotide triphosphate, 10 pmol of each primer, 1 μg of the template DNA, and 1.25 U of Taq DNA polymerase. The PCR was performed as follows: denaturation at 94°C for 30 sec, annealing at 53°C for 30 sec, and extension at 72°C for 90 sec for 30 cycles, with a final extension at 72°C for 10 min. The amplified DNA fragment was cloned into an expression vector, pCold TF, b and the cloned fragment was confirmed by DNA sequencing. Escherichia coli strain JM109 was transformed with the recombinant plasmid and plated onto a lysogeny broth agar plate that contained 100 μg/ml of carbenicillin at 37°C. Expression of recombinant protein was done essentially according to the manufacturer's instructions. The recombinant protein, encompassing the full length of CAEV p55 with an N-terminal histidine tag and a trigger factor, designated rp55gag, was harvested from the soluble fraction of E. coli, purified with a Ni-chelate column, c and concentrated by ultrafiltration. d The relative molecular size and antigenicity of the recombinant protein were confirmed by sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS-PAGE) followed by WB analysis. The SDS-PAGE was performed by the method of Laemmli. Each antigen was diluted with an equal volume of 2 x sample loading buffer (0.1 M Tris—HCl, 4.0% SDS, 20% glycerol, 10% bromphenol blue, 10% β-mercaptoethanol) and loaded to 12.5% of polyacrylamide gel. The proteins were blotted onto a nitrocellulose membrane by using a blotting device, e and the membrane was incubated with blocking buffer (0.6% skim milk in Tris-buffered saline solution with Tween 20 [TBST; 0.02 M Tris, 0.15 M NaCl, 0.05% Tween 20, pH 8.0]) for 1 hr at room temperature. After blocking, the membrane was incubated with serum samples or positive reference serum diluted 1:4,000 in the blocking buffer for 1 hr at room temperature. The membrane was washed 3 times with TBST, and the bound antibody was detected by incubation with horseradish peroxidase—conjugated rabbit anti-goat immunoglobulin G (IgG), f 1:10,000 diluted by diluent buffer (10% Block Ace g in TBST). The reaction was made visible with a peroxidase substrate chromogen. h The SDS-PAGE analysis revealed a protein band of 104.5 kDa, the predicted size of rp55gag (Fig. 1a), and the specificity (Sp) of the recombinant protein was confirmed by CAEV-positive goat serum in WB (Fig. 1b). The negative control protein expressed by the empty pCold TF vector was recognized in SDS-PAGE analysis as a band with a predictable size (53.2 kDa) but was not detected by the CAEV-positive goat serum in WB (Fig. 1A, B).
For use as an antigen for the wELISA and whole virus WB (wWB), CAEV40 was purified from the culture fluid of infected FLL cells by sucrose density gradient centrifugation in accordance with previous studies. 1,14 The purified virus was resuspended in phosphate buffered saline (PBS) solution and treated with 1% Nonidet P40 in TNE (10 mM Tris—HCl, pH 8.0, 100 mM NaCl, 1 mM ethylenediamine tetra-acetic acid) buffer.
Optimal conditions for ELISAs were determined by checkerboard titration, and protein concentrations of each antigen for the ELISAs were determined by the method of Lowry. Each antigen was diluted in carbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.6) to the optimal concentration (rp55gag: 300 ng/ml, CAEV40: 2.66 μg/ml). The procedure for both ELISAs was as follows: 50 μl of diluted antigen was added to each well of 96-well immunoplates. i The plates were incubated overnight at 4°C and washed twice with 0.05% Tween 20 in PBS (PBST) solution by using an automatic washer. j The coated plates were filled with 200 μl of blocking buffer (3% skim milk in PBS solution). The plates then were incubated for 1 hr at 37°C and subsequently washed 4 times with PBST solution. After blocking reaction, 50 μl of 1:400 diluted serum in diluent buffer (0.15% Tween 20, 2% Block Ace, 0.2 mg/ml ovalbumin in PBS solution) was added to the plates. In each plate, 1 positive reference control and 3 negative reference controls (2 negative sera and 1 well lacking serum) were added in triplicate. Sample sera were added in duplicate. The plates were incubated for 1 hr at 37°C and washed 4 times with PBST solution. Subsequently, the plates were filled with 50 μl of 1:40,000 diluted horseradish peroxidase—conjugated anti-goat IgG. After 1 hr of incubation at 37°C and washing, 50 μl of a chromogenic substrate k was added to the plate. The plates were incubated for 30 min at 37°C, and the chromogenic reaction was stopped by acidification by using 50 μl of 1 M phosphoric acid solution. Finally, the optical density of each well was read at 450 nm by using a microplate reader, 1 and mean absorbance was determined for each sample. For the AGID test, a Japanese isolate, the CAEV40 strain, was used as the antigen. The preparation of the antigen and procedure for the AGID test were carried out as previously described. 11,12
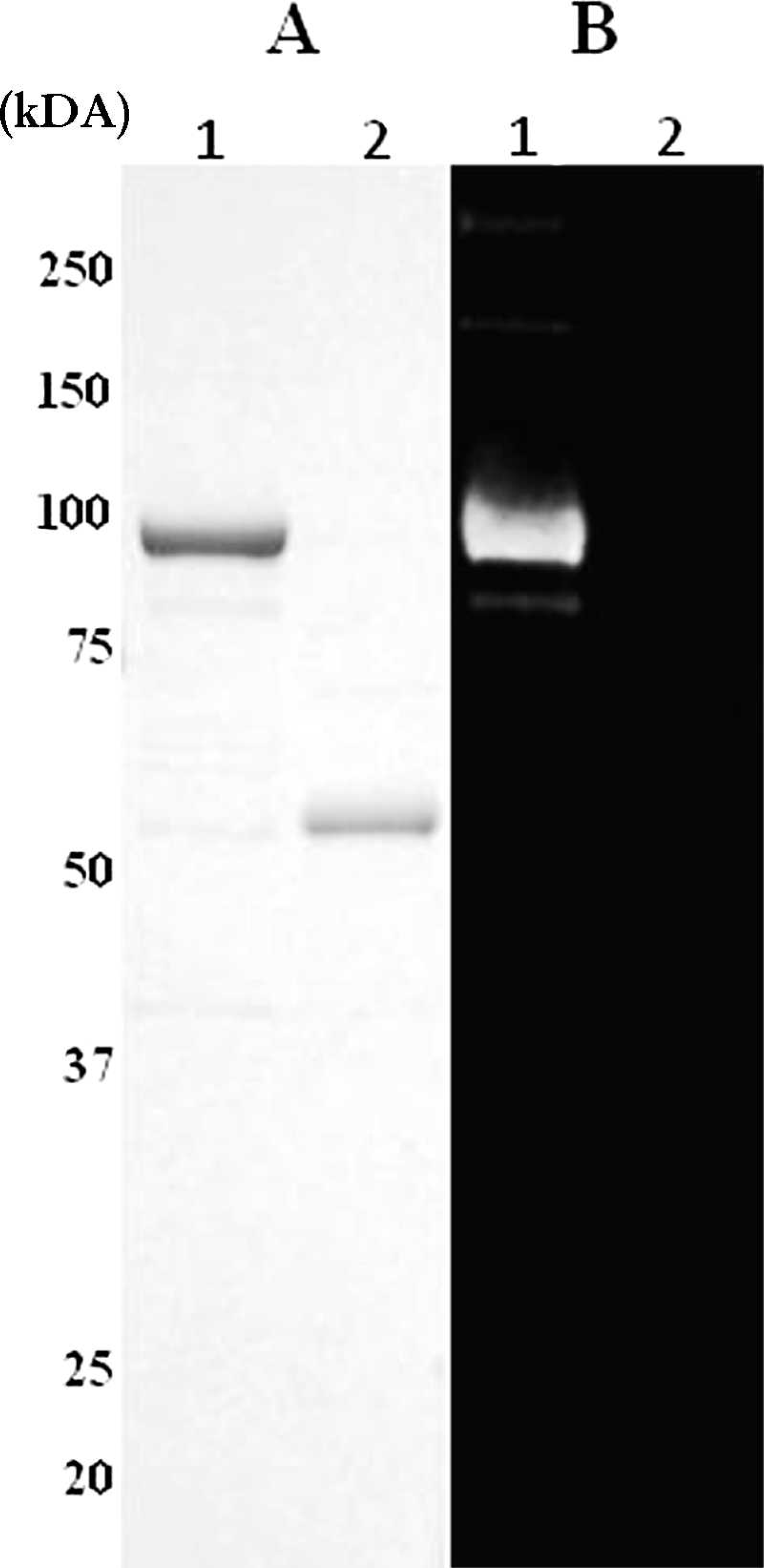
Expression of recombinant p55gag (rp55gag).
To evaluate the performance of the ELISAs, sera of 745 goats collected from 16 farms in 11 prefectures of Japan were tested by the 2 ELISAs, the AGID test, and the 2 WBs. The positive reference serum for each test was obtained from a goat that had clinical symptoms and histopathologic findings of CAE. The goat was also positive in the AGID test and PCR, which was conducted as in a previous study. 11 The negative reference sera were obtained from 2 adult goats that had been constantly monitored serologically and found negative for the previous 2 years. These goats were negative by PCR during the same period. The wWB and rWB were carried out by using the same procedure as the WB analysis described above. A positive sample was defined when at least one of the structural proteins of CAEV, 2,6,8 matrix/nucleocapsid (p16/19), capsid (p28), or an oligomer of the transmembrane protein (gp90) was detected by wWB. In wWB, 372 of the 745 sera were positive (Table 1), and these data were used for reference. The data for each ELISA were subjected to 2-graph receiver operating characteristic (TG-ROC) analysis according to the reference, as shown in Figure 2. In the TG-ROC curve, sensitivity (Se) and Sp were plotted as dependent variables separately against the assumed cutoff value as an independent variable, and the numbers of false positives and false negatives were lowest at the intersection of the 2 curves (Se and Sp). 9,20 Thus, an optical density value of 0.8, which corresponded to the intersection, was selected as the cutoff value for both ELISAs.
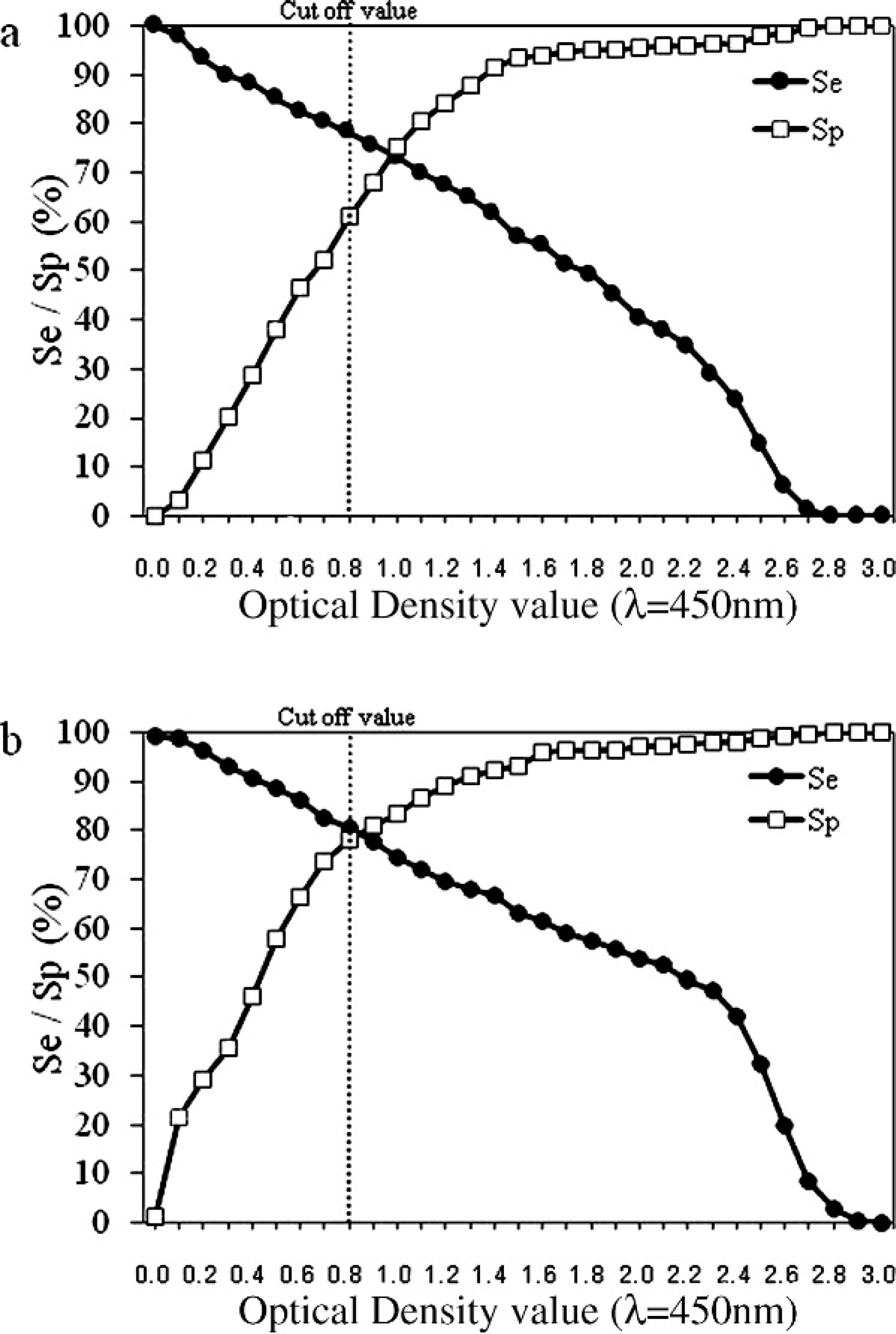
Two-graph receiver operating characteristic (TG-ROC) analysis of each enzyme-linked immunosorbent assay (ELISA); each TG-ROC curve indicates sensitivity (Se) and specificity (Sp) of ELISA at serial optical density values. Dotted line shows cutoff value.
Comparison of results between the Western blot analysis using whole Caprine arthritis encephalitis virus (CAEV) antigen (wWB) and other tests.*
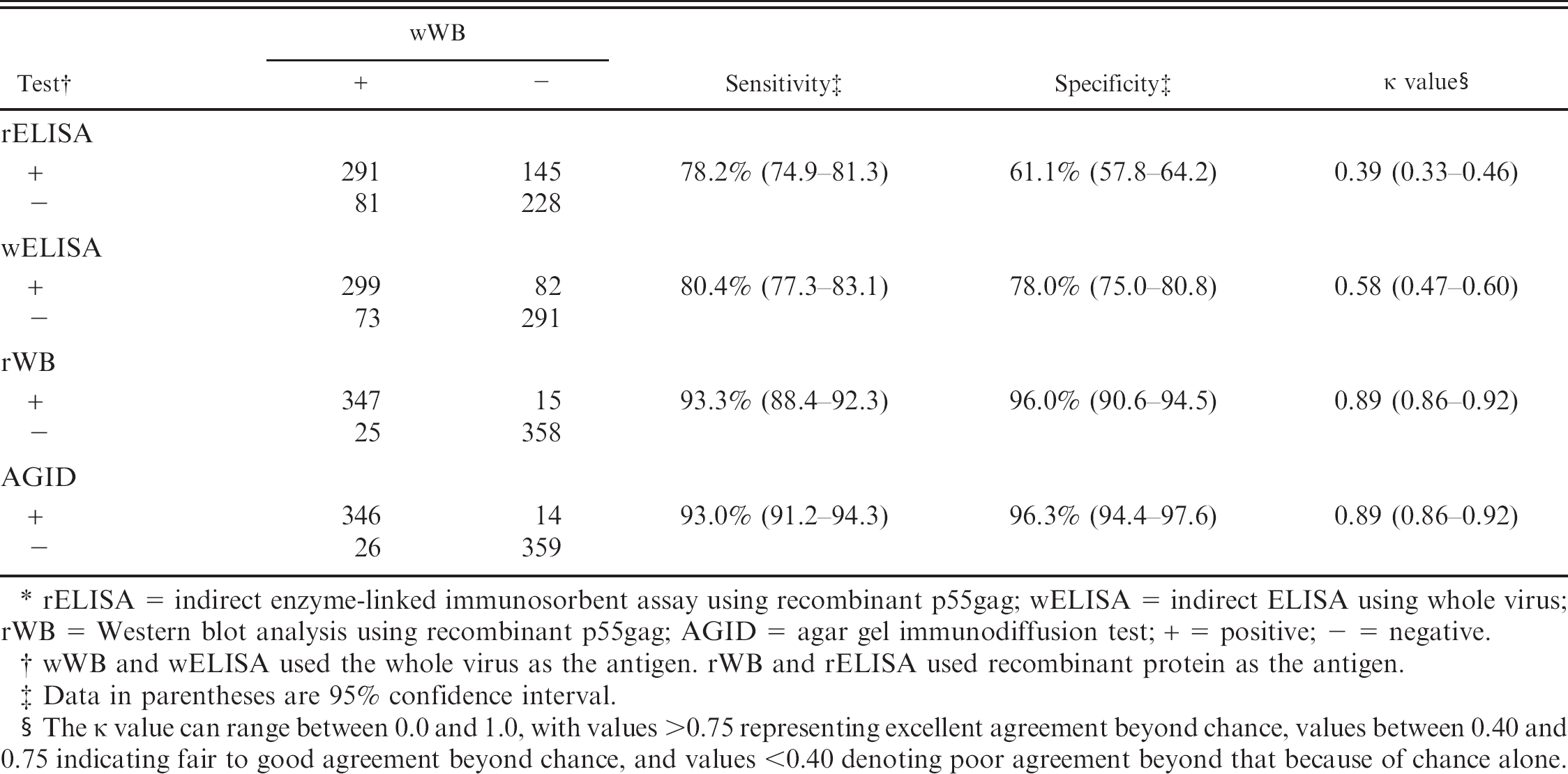
rELISA = indirect enzyme-linked immunosorbent assay using recombinant p55gag; wELISA = indirect ELISA using whole virus; rWB = Western blot analysis using recombinant p55gag; AGID = agar gel immunodiffusion test; + = positive; − = negative.
wWB and wELISA used the whole virus as the antigen. rWB and rELISA used recombinant protein as the antigen.
Data in parentheses are 95% confidence interval.
The κ value can range between 0.0 and 1.0, with values >0.75 representing excellent agreement beyond chance, values between 0.40 and 0.75 indicating fair to good agreement beyond chance, and values <0.40 denoting poor agreement beyond that because of chance alone.
The results of both ELISAs were compared with those of the other tests relative to that of wWB. The results of each test versus wWB are shown in Table 1. The Se, the Sp, and the agreement between wWB and each test, which was evaluated by the kappa (κ) statistic, are also shown in Table 1. Both rWB and the AGID test had higher Se than both ELISAs when using wWB as the standard of comparison.
The CAEV p55gag forms the core proteins p28, p19, and p16, which are major antigens of SRLVs. 6,13,22 The antibodies against the core proteins are raised in most infected animals at the earliest stage of infection, 15,18 and results of previous studies suggest that p55gag of SRLV itself can be recognized by antibodies against each core protein. 2 Thus, rp55gag was presumed to contribute to increasing the opportunity to bind antibodies against CAEV in goat serum, because rp55gag is a multiple antigen. Contrary to the hypothesis, gp90, the oligomer of the transmembrane protein of CAEV reacted with most sera with wWB (data not shown). Results of recent studies suggest that combined usage of the capsid protein and envelope protein, especially the oligomer of transmembrane protein, which are immunodominant in goats with progressive arthritis, is essential for sensitive rELISA. 4,5,10,16,21,23 The results presented herein support those previous studies.
The dilution ratio of goat sera was as high as 1:400, because the negative reference sera showed high background levels at low dilution ratios in both ELISAs. Some of the sample sera might have been judged as false negative because of the high dilution ratio. This could be the reason for the reduced Se of rELISA and wELISA compared with wWB.
The findings that rELISA and wELISA had much lower Sp than the AGID test or rWB suggested that there was nonspecific binding of the antibody in both of these ELISAs. Some possibilities include incomplete blocking steps and inadequate washing in the ELISAs. In addition, sera were diluted 10-fold more in rWB than in rELISA and wELISA, which could have contributed to the overall observed lower Sp of the 2 ELISAs.
In preparation of the antigen, rELISA has an advantage over wELISA because a large amount of a homogeneous antigen can be obtained at one time without large-scale cell culture and virus purification in the assay. However, the rELISA developed in the present study still needs additional work. Combined usage of recombinant envelope protein and rp55gag, and improvement of the reaction system to block nonspecific binding may increase the accuracy of rELISA. However, the results from rWB show that this assay has excellent agreement with wWB. The wWB, by using rp55gag as the antigen, can be used as a confirmatory test for CAEV infection as with other lentivirus.
Acknowledgements The authors thank the Livestock Hygiene Service Center workers for their cooperation in providing blood samples, and Mr. K. Barrymore for his critical reading of the manuscript. This study was partly supported by a special grant from the National Institute of Animal Health, Japan.
Footnotes
a.
TaKaRa Ex Taq®, Takara Bio Inc., Ohtsu, Japan.
b.
pCold® TF, Takara Bio Inc., Ohtsu, Japan.
c.
HisTrap HP, GE Healthcare UK Ltd., Buckinghamshire, UK.
d.
Centriprep® YM-100, Millipore Corp., Bedford, MA.
e.
iBlot Gel Transfer Device, Life Technologies Inc., Gaithersburg, MD.
f.
Rabbit anti-goat IgG, O.E.M Concepts Inc., Saco, ME.
g.
Block Ace, Dainippon Sumitomo Pharma Co. Ltd., Osaka, Japan.
h.
Supersignal® West Dura Extended Duration Substrate, Thermo Scientific, Rockford, IL.
i.
Nunc F96-well microplate (Cert-Maxisorp Immuno plate), Thermo Fisher Scientific Inc., Waltham, MA.
j.
Auto mini-washer AMW-2 plate washer, BioTec Co. Ltd., Tokyo, Japan.
k.
SureBlue Reserve TMB 1-Component Microwell Peroxidase Substrate, Kirkegaard and Perry Laboratories, Gaithersburg, MA.
l.
Immuno Mini NJ-2300, Nalge Nunc International K. K., Tokyo, Japan.