
Abstract
Lawsonia intracellularis, a causative agent of porcine proliferative enteropathy, is an obligate intracellular bacterium that is difficult to culture, propagate, and quantify. The intestinal epithelial cell line (IEC-18, derived from rat small intestine crypt cells) has been used to isolate and propagate this pathogen. However, the lack of rapid and simple quantification methods has led to mixed results when using the rat cell line, complicating Lawsonia studies. To overcome these limitations, a SYBR green quantitative polymerase chain reaction (qPCR) assay targeting a unique hypothetical protein was developed for detecting and quantifying L. intracellularis in IEC-18 rat epithelial cells, porcine fecal samples collected from different farms, and experimentally infected pigs. The method was optimized to detect as few as 3 copies per qPCR reaction of the bacterium growing in IEC-18 cells, providing a new and necessary approach to assess the growth of L. intracellularis in these cell lines. Furthermore, the qPCR assay was successful in detecting L. intracellularis in fecal samples collected from pigs with and without a history of Lawsonia infections, as well as in experimentally infected pigs, which further confirmed the suitability of the qPCR assay for studying the epidemiology of this pathogen.
Lawsonia intracellularis causes proliferative enteropathy, an economically significant gastrointestinal disease in pigs. 7 Lawsonia intracellularis infections are complicated by incomplete or inconsistent data describing the microbiological characteristics and metabolism of this organism. 4 Furthermore, L. intracellularis is an obligate intracellular bacterium that is difficult to culture, 5 and its detection and quantification pose a challenge for epidemiological and infection studies. However, L. intracellularis is commonly propagated in cells from the intestinal epithelial cell line (IEC-18, derived from rat small intestine crypt cells), which is regarded as the most practical approach for the isolation of this pathogen and preparation of infectious inocula for animal studies. 7,10 Because culturing options for L. intracellularis are currently limited, IEC-18 cells are crucial for research and control efforts. However, due in part to the absence of tools that allow rapid quantification of L. intracellularis in these cell lines, researchers have reported varying successes when using these cell lines as an isolation and propagation system. 4,10 Because detection of L. intracellularis infection of pigs that are in different production stages is critical in reducing related losses in food animal production, the purpose of the current study was to develop a SYBR green real-time polymerase chain reaction (real-time PCR) assay for the detection and quantification of L. intracellularis in IEC-18 rat cells and porcine fecal samples.
Because previous researchers reported mixed results for propagating L. intracellularis in rat cell lines, it was important in this study to establish the growth of this pathogen in these cell lines. Lawsonia intracellularis (American Type Culture Collection [ATCC] 55672) was grown in IEC-18 cells (ATCC CRL-1589) as described previously 8 with minor modifications. Phase contrast microscopic observations after serial passages in the IEC-18 cells (data not shown) and quantitative (q)PCR analysis (an average of 15,000 copies/ml were detected after each passage, as described below) indicated the growth of L. intracellularis, which was further confirmed by fluorescent microscopy and 5-cyano-2,3-ditolyl tetrazolium chloride staining, 1 which showed that the bacterium retained its viability in these cell lines (data not shown). Further analysis by electron microscopy showed that the IEC-18 cells harbored curved, mostly nonflagellated L. intracellularis that were approximately 2 μm in length, whereas few cells had a remnant of a flagella-like appendage (data not shown).
For detection of L. intracellularis with a qPCR approach, it is important to use specific and robust primers under optimal conditions. Consequently, qPCR primer sets (Table 1) unique to L. intracellularis were selected after screening against L. intracellularis genome, as well as the entire GenBank database, including closely related Desulfovibrio spp. with the use of BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). To determine the most specific primer set, qPCR was performed on 1 μg of total genomic DNA (gDNA) that was extracted from the Lawsonia-infected IEC-18 cells with a DNA purification kit. a SYBR green qPCR was carried out with the use of a commercial SYBR kit b in a Realplex 2 PCR machine. c Amplification was performed with 300 nM of each primer set with the following conditions: 95°C for 10 min, 40 cycles of 95°C for 15 sec, an annealing temperature gradient (50–62°C) for each primer set for 30 sec, and a 72°C elongation for 30 sec. The SYBR green readings were taken at each 72°C elongation step and after each 0.5°C increase in temperature during a postamplification melt curve. From samples with a single-peak melt curve, which indicated the amplification of a single product, the candidate with the lowest threshold cycle (Ct) value was identified. Two primer sets, LI0028 and PEP-synthase, had multiple peaks in the postamplification melt curve analysis, indicating that these primers were not specific to target DNA. However, both the 16S ribosomal DNA (rDNA) and hypothetical protein LI0045 (HP) primer sets yielded specific products with optimal annealing temperatures of 54°C and 60°C, respectively. Although the 16S rDNA primers had a Ct value 0.69 cycles lower than the HP primer set, the HP primer set was more specific and did not yield any noticeable product from uninfected IEC-18 cells. Alternatively, the 16S rDNA primers resulted in a nonspecific amplification at a Ct value of 30.5 under these conditions. The specificity of the HP primers was further established by sequencing the PCR product (accession no. GU056167), which indicated that the HP primer set was suitable for the detection of L. intracellularis.
List of primers used in the current study. *

Primers for quantitative polymerase chain reaction were designed with the use of Beacon Designer version 7.01 e on the basis of the published genome sequence of Lawsonia intracellularis (GenBank accession NC_008011). Four primer sets were selected to target: the 16S ribosomal RNA gene, gene encoding hypothetical protein, LI0028, and the PEP-synthase/pyruvate phosphate sequence.
Hypothetical protein qPCR specificity was further tested with gDNA from several enteric bacteria, including Campylobacter coli (ATCC 33354), Campylobacter jejuni (National Collection of Type Cultures [NCTC] 11168), Salmonella enterica serovar Typhimurium, d Clostridium difficile (ATCC 9569), Lactobacillus rhamnosus (ATCC 53103), Bifidobacterium animalis subspecies animalis (ATCC 27536), Escherichia coli, d and the closest bacterial relatives to L. intracellularis, Desulfovibrio desulfuricans (ATCC 27774) and Bilophila wadsworthia (ATCC 49260). None of these samples yielded any product even after 45 cycles. Furthermore, analysis of total DNA (representing the bacterial community occurring in the pig's gut), which was extracted from fecal samples of known Lawsonia-negative (n = 5) and gnotobiotic (n = 2) pigs, remained negative even after 45 cycles. These results further indicated that the HP primers were specific to L. intracellularis.
The analytical sensitivity of the HP qPCR was determined by dilution experiments, which affirmed a log-linear relationship between the Ct value and the starting concentration of L. intracellularis in IEC-18 cells (Fig. 1). For each 10-fold change in L. intracellularis, there was an approximate 4-cycle change in Ct value. This indicated an efficient and consistent PCR amplification and qPCR data that reflected the change in L. intracellularis numbers and growth in the IEC-18 cells. On average, the Ct value for positive infection of IEC-18 cells was 33.3 (SD = 3.4) with a detection limit of 3 copies per PCR reaction. A parallel dilution curve of gDNA extracted from different dilutions of Lawsonia-spiked feces demonstrated similar sensitivity, with a detection limit of 6 copies per PCR reaction (Fig. 1). Collectively, these assays showed that HP qPCR was sensitive for detection and quantification of even relatively low numbers of L. intracellularis in fecal samples and IEC-18 cells.
The lack of appropriate quantification tools and the unculturability of L. intracellularis on common growth media have limited the usability of rat cell lines. For example, a previous study 3 reported that coculture of L. intracellularis in rat enterocytes did not yield a sufficient number of bacterial cells, whereas another study 4 did not detect any growth in these cell lines. Therefore, in the current study, an assay was developed to determine the copy number of L. intracellularis in the infected IEC-18 cells. For this purpose, the amplification of the rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene 13 was compared to the amplification of Lawsonia HP. Because the number of IEC-18 cells used for infection was counted by Trypan Blue exclusion and the number of GAPDH was previously determined as 400 copies in the rat genome, 15 the GAPDH Ct value could be correlated to the number of cells. This information was used to determine the approximate number of L. intracellularis by comparing it to the Ct value of HP, which has 1 copy per Lawsonia chromosome (determined by BLAST and genome inspection). Therefore, 1 μg of total gDNA was extracted from an equal number of Lawsonia-infected and -uninfected IEC-18 cells to be used for qPCR, and the number of L. intracellularis was calculated according to the following estimate: approximate Lawsonia concentration (cells/ml) = IEC-18 (cells/ml)/10[HP Ct–(GAPDH Ct+8]/3.3. Because 400 copies of GAPDH are present in the rat genome, 13,15 the Ct value for GAPDH was approximately 8 cycles lower than the HP Ct value. The difference in the GAPDH Ct value, corrected for copy number from the HP Ct value, is a measure of fold change, wherein approximately every 3.3 cycles is equal to a 10-fold change. Lawsonia-infected IEC-18 cells or intestinal homogenates are commonly used as challenge material to determine effectiveness of therapeutic and bio-security measures. 2 To demonstrate the importance of the qPCR assay, 5 Lawsonia-free grower pigs (4 weeks old) were experimentally inoculated with L. intracellularis grown in IEC-18 cells (5 × 105 bacteria per pig), as described previously. 10 Two of the pigs were euthanatized on day 21 postinoculation and showed gross postmortem lesions that were diagnostic for Lawsonia infection. 6 Interestingly, disease symptoms corresponded with relatively higher fecal shedding of Lawsonia at day 21 compared with the earliest time point, which also indicated that the IEC-18 inoculum containing L. intracellularis resulted in the successful colonization of the pigs (Fig. 2). Regardless, these experiments were designed to show the usefulness of the qPCR assay and the importance of quantifying L. intracellularis in IEC-18 cells, and the results confirmed that the method provided a means to monitor Lawsonia shedding from experimentally infected pigs.
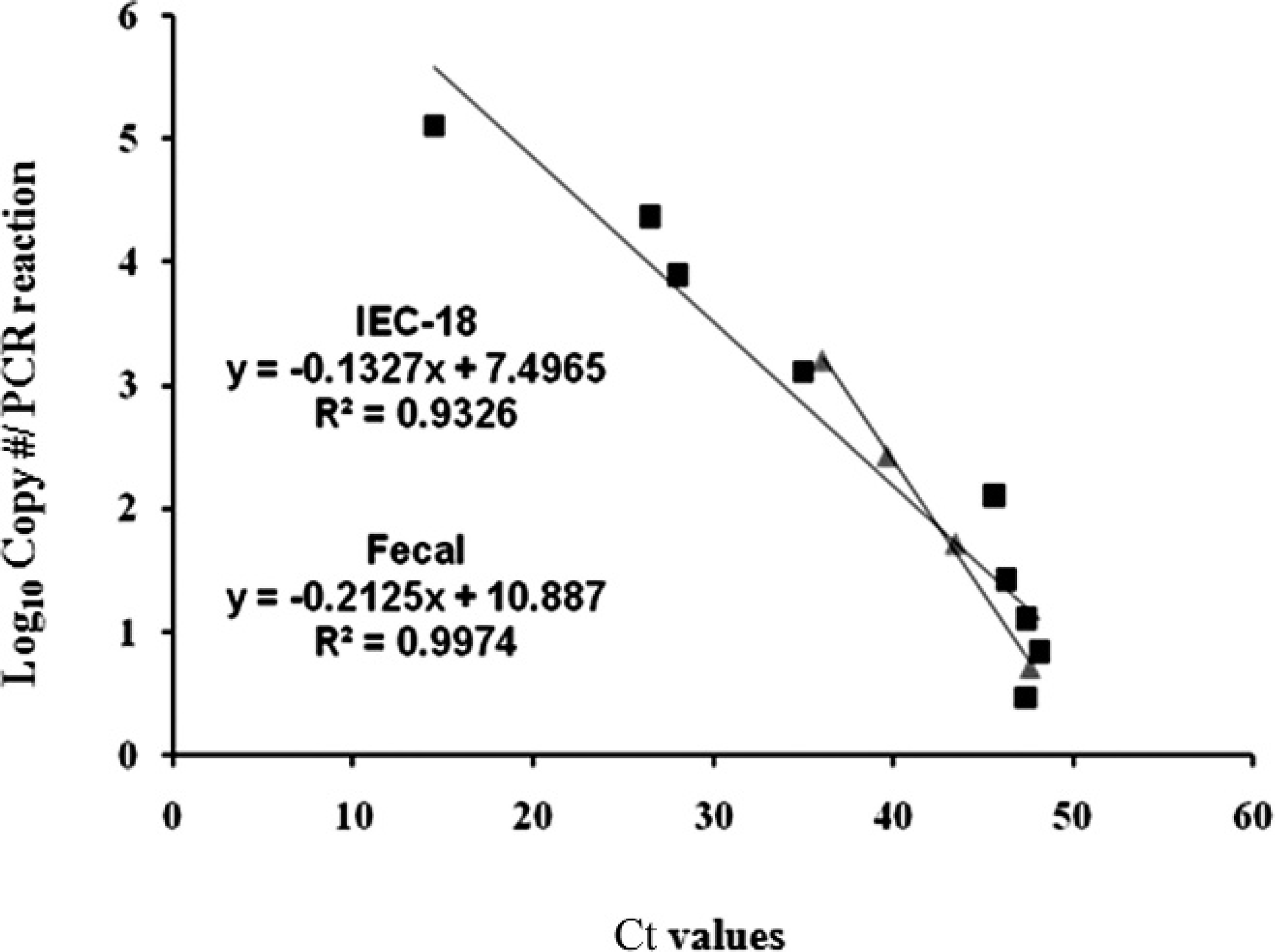
Relationship between the threshold cycle (Ct) values and the Lawsonia intracellularis copy number. Different dilutions of DNA extracted from L. intracellularis–infected IEC-18 cells (▪) or feces (▴) were tested in 2 independent experiments, and each sample was run in triplicate to determine the detection limit. Analytical sensitivity was tested by serial dilutions (10-fold) of IEC-18 cells or feces containing a prequantified number of L. intracellularis.
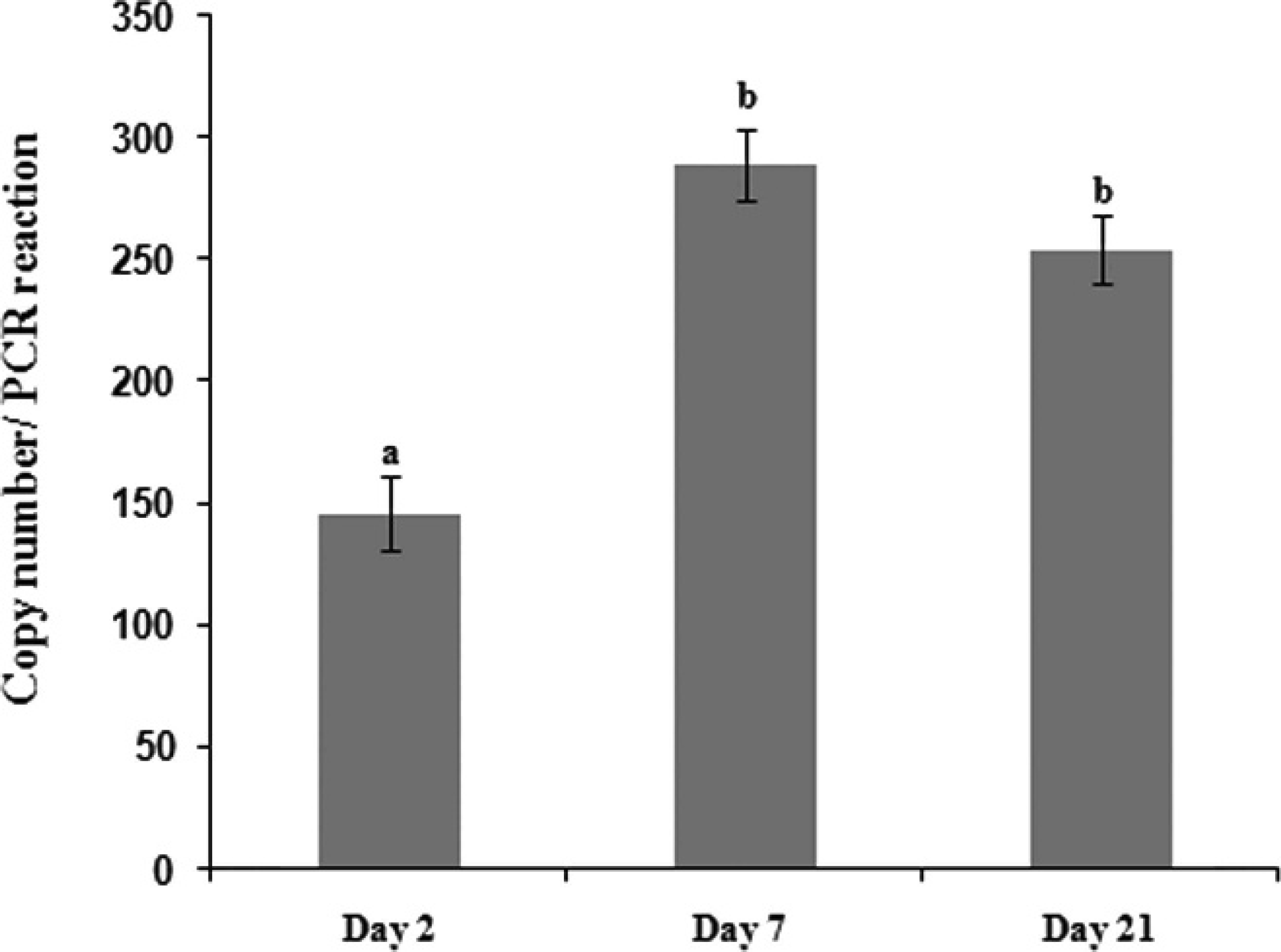
Quantitative polymerase chain reaction (qPCR) of genomic DNA extracted from fecal samples that were collected from experimentally infected 4-week-old grower pigs (n = 5). Fecal samples were analyzed by hypothetical protein qPCR in 2 independent experiments, and each sample was run in triplicate to determine Lawsonia copy number. Data represent geometric mean ± standard error. Different letters highlight significantly different means (P < 0.05).
To test whether the SYBR green qPCR assay would be useful for the detection of L. intracellularis in potentially infected pigs, fecal matter was collected from 3 different farms. Farm E (pigs 1–10), which had a history of endemic L. intracellularis infection and ileitis cases; farm O (pigs 11–15), with unknown Lawsonia status; and farm W, with known Lawsonia-negative status. In addition, samples from 2 gnotobiotic pigs were also examined. Quantitative PCR conditions were optimized for fecal samples that were spiked with tissue culture–grown L. intracellularis by testing a range of annealing temperatures (50–62°C) to identify optimal conditions that yielded a single-peak melt curve with the lowest Ct value, indicating a specific and sensitive amplification. The optimal conditions were initial denaturing at 95°C for 10 min followed by 45 cycles of 95°C for 15 sec, 53.7°C for 30 sec, and 72°C for 30 sec. To test whether fecal-PCR inhibitors were affecting PCR amplification, 100 μl of IEC-18 cells containing 103 Lawsonia cells were pelleted and resuspended in 1 ml of phosphate buffered saline and added blindly to fecal samples from farm E before testing the samples by HP qPCR. All spiked samples were positive by qPCR and had an average increase of approximately 500 Lawsonia copies compared with nonspiked samples from the same pig (Fig. 3). Although approximately the same amount of fecal sample was used for gDNA extraction, small variations in fecal samples as well as nonfecal inclusions (e.g., undigested food material) might be responsible for some of the differences. This was further confirmed when the universal 16S rRNA gene primers 11 that were used as controls for qPCR analysis of DNA from homogeneous fecal matter samples showed consistent amplification across samples (data not shown). For example, universal 16S rRNA gene qPCR for all samples with approximately 30 ng of extracted DNA showed a Ct value of 18 cycles (SD = 1.7). The HP qPCR assay showed that all 10 fecal samples (100%) from farm E were positive, however only 3 out of 5 fecal samples (60%) from farm O were positive (Fig. 3). Additionally, all samples from farm W (n = 5) and those from the gnotobiotic pigs (n = 2) were confirmed negative.
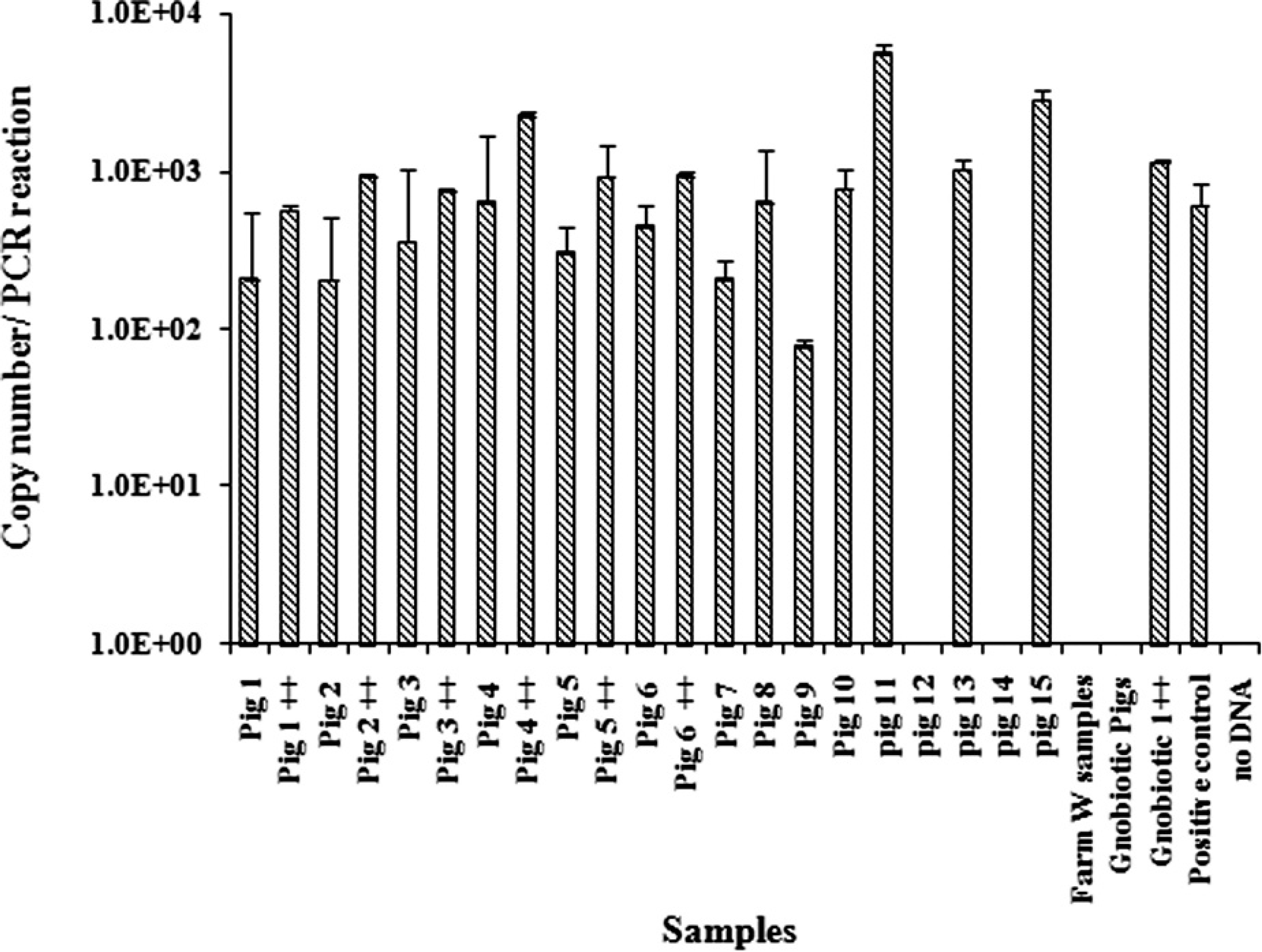
Quantitative polymerase chain reaction of genomic DNA extracted from fecal samples that were collected from farm E (pigs 1–10), farm O (pigs 11–15), and farm W (Lawsonia-negative pigs; n = 5). Genomic DNA samples extracted from Lawsonia-infected IEC-18 cells and gnotobiotic pig feces (n = 2) were used as positive and negative controls, respectively. ++ = fecal samples spiked with Lawsonia-infected IEC-18 culture. Data represent geometric mean ± standard error from 2 replicate runs.
Positive fecal samples had an average Ct value of 38.8 (SD = 1.0) from farm E, an estimated concentration of 309 Lawsonia per extraction and a Ct value of 35.2 (SD = 1.7), with 2,500 Lawsonia per extraction from farm O samples. The average Ct value was higher than expected, which indicated that fecal shedding can be low even in endemically infected pigs. These results, although concerned with a limited sample size, were consistent with the reported prevalence of L. intracellularis in pig herds. 4 Because the DNA from L. intracellularis–negative fecal samples were successfully amplified with the universal 16S rRNA gene primers, L. intracellularis–negative results were not considered a consequence of fecal inhibition.
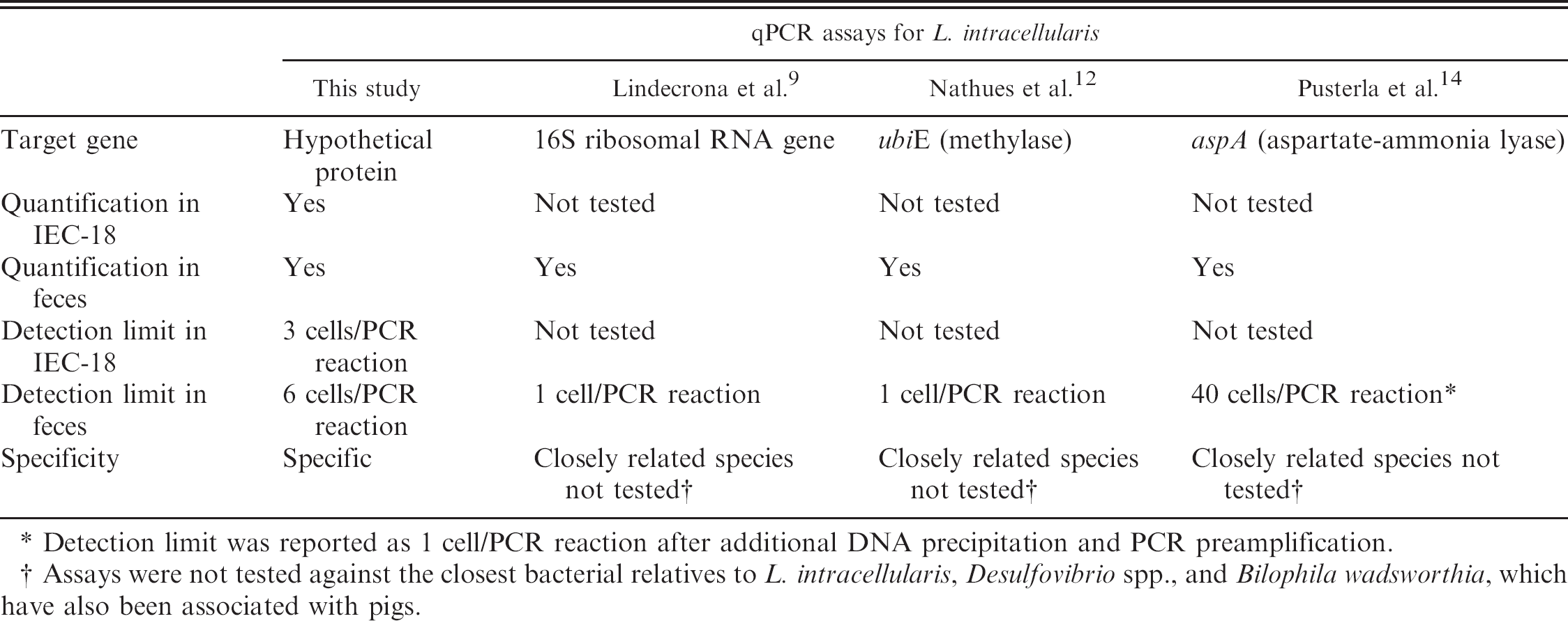
Comparison between quantitative polymerase chain reaction (qPCR) assays used for the detection and quantification of Lawsonia intracellularis.
Detection limit was reported as 1 cell/PCR reaction after additional DNA precipitation and PCR preamplification.
Assays were not tested against the closest bacterial relatives to L. intracellularis, Desulfovibrio spp., and Bilophila wadsworthia, which have also been associated with pigs.
Although TaqMan probe–based real-time assays for the detection of Lawsonia have been recently available, 9,12,14 the assay described in the current study has the advantages of simplicity and relatively lower cost, making it easier to adapt to diagnostic needs required for processing large sample numbers that are commonly associated with epidemiological studies. In contrast to the present study, the TaqMan probe–based assays were not validated by monitoring experimentally infected live pigs and were not tested against Desulfovibrio spp. and B. wadsworthia, the closest bacterial relatives to L. intracellularis. Unlike the HP primers used in the current study, a BLAST analysis of the primers and probe used in a previous qPCR study 9 showed up to 100% similarity to Desulfovibrio spp. (accession no. U07570.1) and low similarity to B. wadsworthia (accession no. U82813.1), whereas in another study, 12 16 bases of the 28-base probe were 100% similar to a sequence in D. desulfuricans (ATCC 27774). Additionally, the HP-qPCR provides a novel approach to quantify Lawsonia in IEC-18 cells with the use of rat GAPDH as an indicator, which has not been performed with other available qPCR assays (Table 2).
Many L. intracellularis cultures are patented items, 4 which coupled with their unculturability on common growth media, might lead to inconsistencies and confusion in studying this bacterium. The method outlined in the current study has several advantages: it 1) is both sensitive and specific, detecting as few as 6 bacterial copies per PCR reaction with the use of DNA extracted from 30 mg of feces and as few as 3 copies per PCR reaction with the use of DNA extracted from 1 ml of L. intracellularis–infected IEC-18 cells; 2) uses SYBR green, yielding a robust, effective, and simple qPCR system that does not require fluorophore-containing probes; and 3) allows the evaluation of successful L. intracellularis growth in IEC-18 cells, which provide suitable conditions for the isolation and propagation of this organism. 10 Finally, the qPCR assay can also identify Lawsonia in fecal samples from suspected equine proliferative enteropathy cases (data not shown), thus making it widely useful for detection of L. intracellularis in multiple species.
Acknowledgements. The authors thank Dr. Tea Meulia for assistance with electron microscopy and the Ohio Agricultural Research and Development Center (OARDC), The Ohio State University, for financial assistance. Research in Dr. Rajashekara's laboratory is also supported by USDA grant 2007–03109.
Footnotes
a.
MasterPure™ DNA Purification Kit, EPICENTRE Biotechnologies, Madison, WI.
b.
SensiMix™ Plus, Quantace Ltd., Norwood, MA.
c.
Eppendorf, Westbury, NY.
d.
Caliper Life Sciences, Hopkinton, MA.
e.
PREMIER Biosoft International, Palo Alto, CA.