
Abstract
Real-time polymerase chain reaction (PCR) with TaqMan minor groove binder (MGB) probes was examined to establish a rapid and reliable genotyping technique for GM1 gangliosidosis in Shiba dogs. This technique was applied to DNA samples extracted from the blood, umbilical cord, or postmortem liver tissue specimens, and to DNA-containing solutions prepared from blood and saliva that had been applied to Flinders Technology Associates filter papers (FTA cards). The amplification of the targeted sequence in all the samples was sufficient to determine the genotypes of GM1 gangliosidosis. Forty-seven DNA samples that had previously been obtained from blood or tissue specimens of Shiba dogs were examined using this real-time PCR technique, and the findings were consistent with the data obtained by the earlier PCR–restriction fragment length polymorphism (RFLP) assay. In addition, the use of this new technique in combination with FTA cards for sampling could markedly shorten the time required for genotyping, as well as simplify the procedure. Furthermore, in the present study, the results of a previous epidemiological screening of 96 Shiba dogs in the Czech Republic were rechecked by this real-time PCR technique using stored crude buccal cell DNA-containing solutions directly as DNA templates. The results provided clear-cut genotyping in all the samples although the earlier PCR-RFLP assay could not determine the genotype in all cases. In conclusion, this new real-time PCR technique is a simple, rapid, and reliable choice for large-scale screening to detect an abnormal allele indicating GM1 gangliosidosis in Shiba dogs.
GM1 gangliosidosis, a lysosomal storage disease that affects the brain and multiple systemic organs, is due to an autosomal recessively inherited deficiency of acid β-galactosidase encoded by the GLB1 gene. 4 GM1 gangliosidosis in Shiba dogs was originally reported in 2000. 13 The causative mutation has already been identified as a deletion of C nucleotide 1647 in the putative coding region for the canine β-galactosidase gene, 7 allowing antemortem diagnosis with a polymerase chain reaction (PCR)-based DNA test 11,12 in addition to biochemical diagnosis with an enzyme assay using leukocyte or tissue specimens. 10,12
The Shiba dog is a popular canine breed originating in Japan and has been designated a protected species in that country. Approximately 40,000 puppies are produced and registered every year in Japan. 9 Moreover, Shiba dogs have been transported all over the world, and are bred and maintained as a standard breed in many countries. 9 Therefore, it is very important to determine the genotypes of Shiba dogs for GM1 gangliosidosis to control this fatal inherited disease.
Recently, rapid real-time quantitative PCR approaches have been developed to detect mutations in genes causing hereditary diseases. 2,5,6 Real-time PCR is generally considered more sensitive, rapid, and less time consuming than conventional PCR for the analysis of genetic variability. In the present study, therefore, the application of real-time PCR with TaqMan minor groove binder (MGB) probes a was investigated to establish a rapid and reliable genotyping technique for GM1 gangliosidosis in Shiba dogs. In addition, the use of Flinders Technology Associates filter papers b (FTA cards) as an alternative to collection and storage of blood or saliva samples in tubes was also evaluated to simplify the procedure and shorten the time required for genotyping.
DNA samples were prepared in different ways. Blood, umbilical cord, or postmortem liver specimens were collected from 47 pedigreed Shiba dogs of families with GM1 gangliosidosis; genomic DNA samples were isolated from these samples using a commercial kit c according to the manufacturer's direction. The genotypes had already been determined by a previously reported PCR–restriction fragment length polymorphism (RFLP) assay. 7,12 To evaluate the usefulness of FTA cards, whole blood or saliva was spotted onto an FTA card, dried at room temperature, and stored at approximately 4°C until used. A 1.2-mm diameter disk was punched out of the FTA card using a hole punch. b The disk was placed into a separate 0.5-ml tube, lysed in the tube with 8 μl of lysis solution, a and subsequently incubated at 95°C for 3 min. Then, 8 μl of DNA stabilizing solution a was added to the tube. This DNA-containing solution was transferred to a new tube and stored at −25°C until analysis. Furthermore, in the present study, the results of a previous epidemiological screening in 96 Shiba dogs in the Czech Republic were rechecked by this real-time PCR technique using crude buccal cell DNA-containing solutions directly as DNA templates, which had been collected in 2005–2006 using a commercial kit d and stored at −25°C. Genotypes in all cases except one had been determined using a previously reported PCR-RFLP assay. 8
Characteristics of primers and TaqMan probes used in the current study. ⊃

NA = not applicable; VIC = 6-carboxyrhodamine; FAM = 6-carboxyfluorescein; NFQ = nonfluorescent quencher.
Amplifications were performed on a real-time PCR system a using TaqMan MGB probes and a specific primer pair. The primers and probes were synthesized by Applied Biosystems, and their sequences and reporter and quencher dyes are shown in Table 1. Concentrations of purified DNA samples were determined spectrophotometrically and adjusted to 20–50 ng/μl. The real-time PCR amplifications were carried out in a final volume of 10 μl consisting of TaqMan master mix (2×), a TaqMan genotyping assay mix (80×) a including specific primers and TaqMan MGB probes, nuclease-free water, and 2 μl of template DNA. A negative control containing nuclease-free water instead of DNA was included in each run. The cycling conditions were 20 sec at 95°C followed by 40 or 50 cycles of 3 sec at 95°C and 20 sec at 61°C. The total required time for 40-cycle amplifications was within 40 min.
DNA extracted from blood, umbilical cord, or postmortem liver tissue specimens as well as DNA-containing solutions prepared from blood or saliva on FTA cards were suitable for clearly determining the genotype of GM1 gangliosidosis by the real-time PCR described in the current study, although DNA-containing solutions prepared from blood or saliva on FTA cards needed more PCR cycles for adequate amplifications compared with that for the samples extracted and purified from blood or tissue specimens (Fig. 1). However, 50 cycles of amplification were sufficient for genotyping, even in the case of saliva DNA samples on FTA cards, which showed the weakest amplification among the samples tested (Fig. 1B). Considering the low concentration of DNA from saliva applied to an FTA card, the number of punched disks was increased but the amplification capacity was not greatly improved. When 3 disks were used for analysis, no or weak amplification was shown (data not shown). In the present study, when FTA cards were used for sampling, the DNA preparation procedure consisted of only lysis and stabilization processes without washing and purification. Therefore, weak amplification using FTA cards seemed to have been the result of some PCR inhibitors inherent to the sample or derived from the FTA card itself. In the real-time PCR assay, to achieve adequate amplification using DNA prepared from FTA cards, increasing the number of cycles could be a better option than increasing the number of punched disks.
As shown in Figure 1A, nonspecific allelic amplification was observed in the sample with a normal genotype in which false mutant-allelic amplification was slightly increased together with true wild-allelic amplification. These allelic discrimination assays (genotyping) are based on end point data for qualitative assays and adopt MGB probes, which are highly specific, especially for single base mismatches. Therefore, the procedure is useful for single nucleotide polymorphism detection and allelic discrimination. 3 However, in the present study, slight nonspecific amplification was observed, although there was no problem in discriminating allelic genotypes, and a slight improvement in specificity was achieved by increasing the annealing temperature from 60°C to 61°C. In normal genotypes, a wild allele starts to amplify faster than a mutant allele, whereas the 2 alleles start to amplify at the same time in carrier genotypes (Fig. 1A, B), making it easy to discriminate these 2 genotypes.
An allelic discrimination plot was made based on the 3 types of amplification plots (Fig. 2). The data were calculated using software a based on the results obtained using DNA samples from blood or tissue specimens in 47 pedigreed Shiba dogs of families demonstrating GM1 gangliosidosis. Three genotypes were clearly determined by 40 cycles of real-time PCR in all specimens, and the results were completely consistent with those of an earlier PCR-RFLP assay. 7,12 In addition, relatively crude buccal cell DNA lysates from 96 Shiba dogs in the Czech Republic were also successfully analyzed as direct template DNA for the newly developed real-time PCR method. All of their genotypes could be determined clearly (data not shown), although the previous study showed that the earlier PCR-RFLP assay could not analyze 1 of the 96 samples because of a fault in the PCR processing. 8
Consequently, the results described herein verified that real-time allelic discrimination PCR is a rapid and reliable diagnostic tool for GM1 gangliosidosis in Shiba dogs. In recessive inherited diseases, such as lysosomal diseases, carriers that have 1 abnormal allele in the gene pair, but are normal in clinical appearance, are the most important genotype because there are no physical clues to the presence of disease in these dogs, but the mutation is transmitted to half of their progeny. 1 The frequency of carriers in a population substantially exceeds the incidence of affected individuals and thus the carrier state makes recessive disease the most dangerous of all patterns of inheritance in purebred animals. 8 Therefore, to prevent and eradicate this fatal hereditary disease, the determination of the genotypes through systematic monitoring surveys and continuous removal of carriers from breeding colonies might be one of the most important and efficient measures. In the present study, the amplification of DNA prepared from a small amount of blood and saliva applied to FTA cards showed good results for genotyping of this disease. In addition, the use of FTA cards for sampling eliminates the need for traditional multistep extraction and purification procedures, allowing for early reporting of the results within 2 hr after sample collection in combination with the real-time PCR technique. In particular, a clear-cut result obtained from a small amount of saliva on an FTA card would be of the greatest benefit for sampling from many Shiba dogs, as this breed is very nervous and quick tempered compared with other European breed dogs.
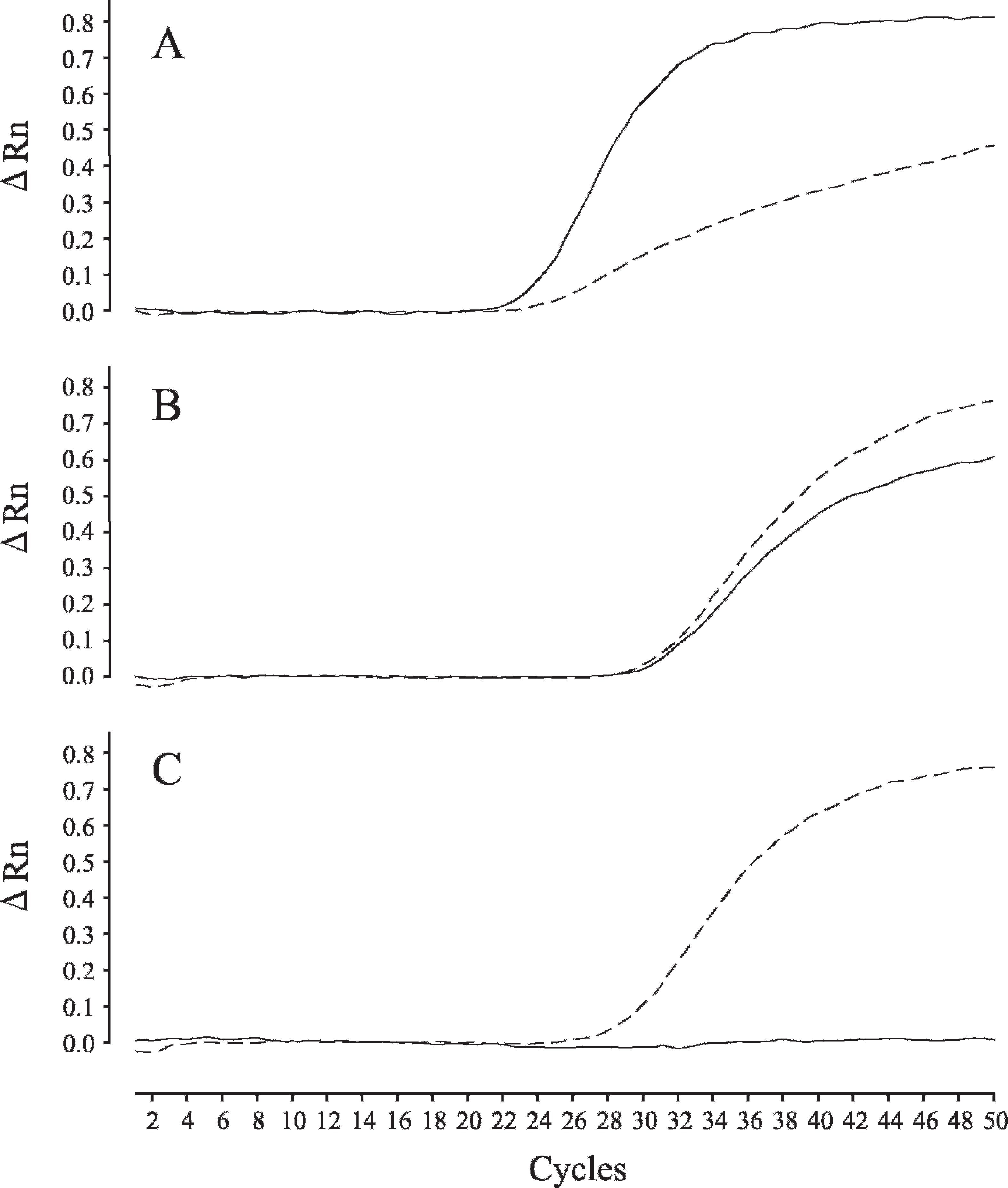
Real-time polymerase chain reaction (PCR) amplification plots of wild- and mutant-type alleles in GM1 gangliosidosis in Shiba dogs. Amplification was plotted as fluorescence intensity (ΔRn value) against cycle number. The ΔRn value is the reporter dye signal normalized to the internal reference dye and corrected for the baseline signal established in the first few cycles of PCR. Each of 3 amplification plots showed the normal genotype (
In conclusion, the current study suggests that real-time PCR is a simple, rapid, and reliable genotyping technique for GM1 gangliosidosis in Shiba dogs. Moreover, this technique, in combination with FTA cards for sampling, can markedly shorten the time required for genotyping and simplify the procedure, which can be particularly useful for large-scale preventive screening, as well as for clinical diagnosis.
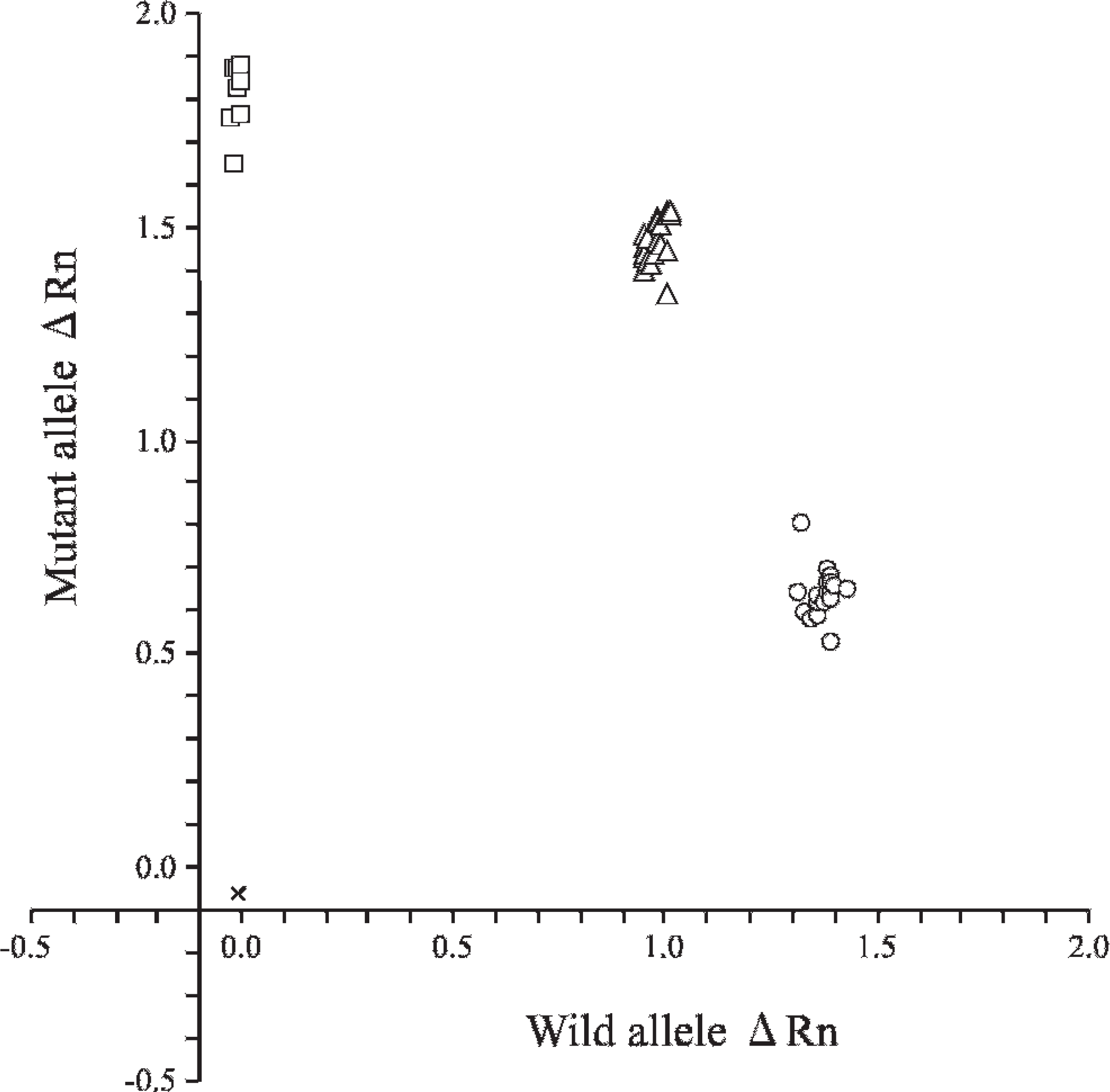
Allelic discrimination plot of end point fluorescence polymerase chain reaction (PCR) data showing the 3 genotypes of GM1 gangliosidosis in Shiba dogs. Allelic discrimination plot was depicted using representative 47 purified DNA samples in pedigreed Shiba dogs of families with GM1 ganglio-sidosis. The plot is expressed as fluorescence intensities (ΔRn values) for each allele at the X- and Y-axes. The ΔRn value in this figure is the end point reporter dye signal normalized to the internal reference dye and corrected for the baseline signal established in the first few cycles of PCR. × = no template control; ○ = normal genotype (18 samples); Δ = carrier genotype (21 samples); □ = affected genotype (8 samples).
Acknowledgements. The authors are grateful to all members of the Shiba Club of the Czech Republic for permission and commitment to the analysis in this study. This study was supported financially by grants (20380173, 20-08112, and 21658109, O.Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) and by the Strategic Research Base Development Program for Private Universities, 2008–2012 (T.A.), which is also funded by MEXT.
Footnotes
a.
TaqMan® MGB™ Probes, DNA Extract All Lysis Reagents Kit, StepOne™ Real-Time PCR System, TaqMan® GTXpress™ Master Mix (2x), TaqMan® SNP Genotyping Assay Mix (80x), StepOne™ software (version 2.0); Applied Biosystems, Foster City, CA.
b.
FTA® classic card, Harris Uni-Core Punch (size 1.2 mm); Whatman International Ltd, Piscataway, NJ.
c.
Easy-DNA™ Kit, Invitrogen Corp., Carlsbad, CA.
d.
BuccalAmp™ DNA Extraction Kit, EPICENTRE Biotechnologies, Madison, WI.