
Abstract
Epizootic hemorrhagic disease virus (EHDV) has been associated with bluetongue-like disease in cattle. Although U.S. EHDV strains have not been experimentally proven to cause disease in cattle, there is serologic evidence of infection. Differentiation of Bluetongue virus (BTV) and EHDV is necessary because diagnosis of infection caused by these viruses is often confused. The previously developed nested reverse transcription polymerase chain reaction (nRT-PCR) test for indigenous EHDV disease is sensitive and specific, but it is prone to contamination problems. Additionally, the EHDV nRT-PCR only detects 7 of the 8 serotypes. To develop an improved diagnostic test, sequence analysis was performed on 2 conserved target genes; one is highly expressed in infected mammalian cells, whereas the other is highly expressed in infected insect cells. This information was used to develop a rapid EHDV real-time PCR that detects all 8 EHDV serotypes. The EHDV assay did not cross-react with BTV strains and performed similarly to the nRT-PCR tests with archived clinical samples. In addition, it is superior to the nRT-PCR, not only because it is a closed system with fewer cross-contamination problems, but also because it detects all 8 serotypes and is less labor and time intensive.
Epizootic hemorrhagic disease virus (EHDV; family Reoviridae, genus Orbivirus) is an insect-transmitted pathogen of ruminants causing periodic significant losses in wild and captive deer populations. 21 Epizootic hemorrhagic disease virus is closely related to Bluetongue virus (BTV), which can also cause hemorrhagic disease in deer. 11 The seroprevalence of BTV and EHDV ranges from 45–100% in deer populations. 6,8 BTV causes an estimated $120 million in annual loss to the U.S. livestock industry and about $3 billion in annual losses worldwide (Bath GF: 1989, Bluetongue. In: Proceedings of the 2nd International Congress for Sheep Veterinarians, pp. 349–357). A clinically similar disease caused by BTV has been associated with EHDV infection in cattle 4,9 ; however, U.S. serotypes of EHDV have not been shown to cause clinical disease in experimental infections. 1 There is also serologic evidence of infection in and isolation of EHDV from U.S. cattle and sheep. 14,23 Therefore, rapid diagnosis and differentiation of BTV and EHDV are needed for both livestock and wildlife.
An understanding of the molecular attributes of these viruses is necessary to develop effective gene-based diagnostic tests. The genome for these viruses comprises 10 segments of double-stranded RNA, which encode for 7 structural and 4 nonstructural proteins. 13,24 There are 24 serotypes of BTV and 8 confirmed serotypes of EHDV. 11,25 Serotype is determined primarily by the outer capsid protein VP2, but it can also be influenced by the second outer capsid protein VP5. 17,29 The inner core proteins VP3 and VP7 are fairly conserved within the virus serogroup. 15,27,31 Nonserotype-specific enzyme-linked immunosorbent assays (ELISAs) for these viruses are usually based on the VP7 protein. 16 Nonstructural proteins of EHDV are highly conserved among North American viruses. 18,19,28 This information has been used to develop rapid genetic amplification tests for early detection and differentiation of indigenous strains of EHDV. 2,28 These previously developed assays all required gel electrophoresis for amplicon detection; therefore, they are time consuming and prone to sample contamination problems. To avoid these problems, a previously described real-time reverse transcription polymerase chain reaction (real-time RT-PCR) assay was developed for EHDV based on sequence data available for the 2 U.S. serotypes of EHDV. 32 However, this assay did not detect all 8 known serotypes of EHDV.
The recent isolations of an exotic serotype of EHDV in the United States (Pearson JE, Wilson WC, Barber TL, et al.: 2007, Report of the Committee on Bluetongue and Bovine Retroviruses. U.S. Animal Health Association. Available at http://www.usaha.org/committees/reports/2007/report-btbr-2007.pdf. Accessed November 21, 2008) clearly demonstrate the need for an improved assay that rapidly detects all known strains. Two genes that are conserved among U.S. serotypes 10,28 were chosen as target amplicons. One gene is highly expressed in infected mammalian cells, segment M5, which encodes nonstructural protein 1 (NS1), 24 and the other is highly expressed in infected insect cells, segment S10, which encodes nonstructural protein 3 (NS3). 5 Sequence analysis of the remaining EHDV serotypes provided the foundation for developing a more robust diagnostic test.
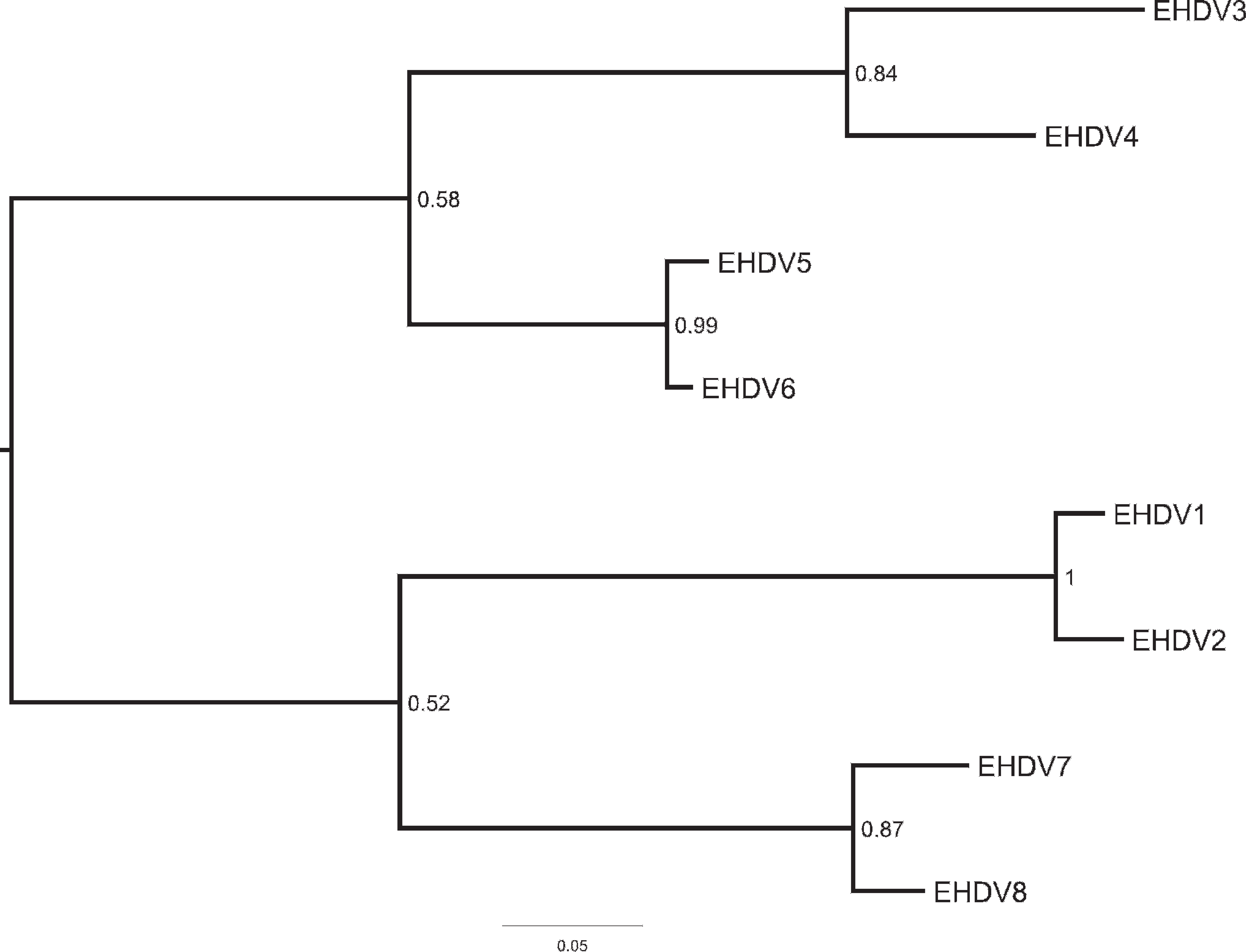
Phylogenetic analysis of the M5 gene from prototype Epizootic hemorrhagic disease virus (EHDV) serotypes. Sequences were aligned using MUSCLE, and the tree was calculated using MrBayes (750,000 generations, GRT+Γ+Inv). Numbers on nodes indicate posterior probabilities. Branch lengths are in nucleotide changes per site.
The EHDV-3 through EHDV-8 prototype viruses were kindly provided by the National Veterinary Services Laboratories. Total RNA was extracted from cells using a commercial extraction kit a per the manufacturer's protocol. Ethanol precipitation was omitted, and the double-stranded RNA (dsRNA) was purified using lithium chloride differential precipitation as previously described. 26 Amplification and sequencing using standard protocols d of the M5 and S10 genes from the exotic EHDV prototype strains were performed as previously described. 3,29 The primer sequences, except for the first 5 bases on the 5′ end that included the ATG start codon for the S10, were removed for data analysis and reported to GenBank (accession nos. EU928893–EU928904). The EHDV M5 gene had a percent identity range from 77.7% (serotypes 4–6 or 7) to 98.6% (serotypes 1–2). The EHDV S10 gene had a percent identity range from 77.2% (serotypes 1–3) to 96.5% (serotypes 1–2). Full-length sequences were aligned using either ClustalX 22 or MUSCLE. 7 Phylogenetic trees were calculated from these multiple sequence alignments using MrBayes 20 using the standard nucleotide substitution model (4 × 4) with GTR (nst = 6). Substitution rates were set to invgamma (gamma-shaped rate variation with a proportion of invariable sites). Default values were used for all other settings. A total of 750,000 generations were calculated by sampling every 100th tree, and a consensus tree was calculated after a burn-in of 2,500 trees using the allcompat setting. The EHDV M5 separated into 2 major lineages with a very high degree of confidence. Phylogenetic analyses of M5 and S10 were performed using several algorithms showing similar profiles regardless of the algorithm but distinct by gene segment (Figs. 1 and 2, respectively).
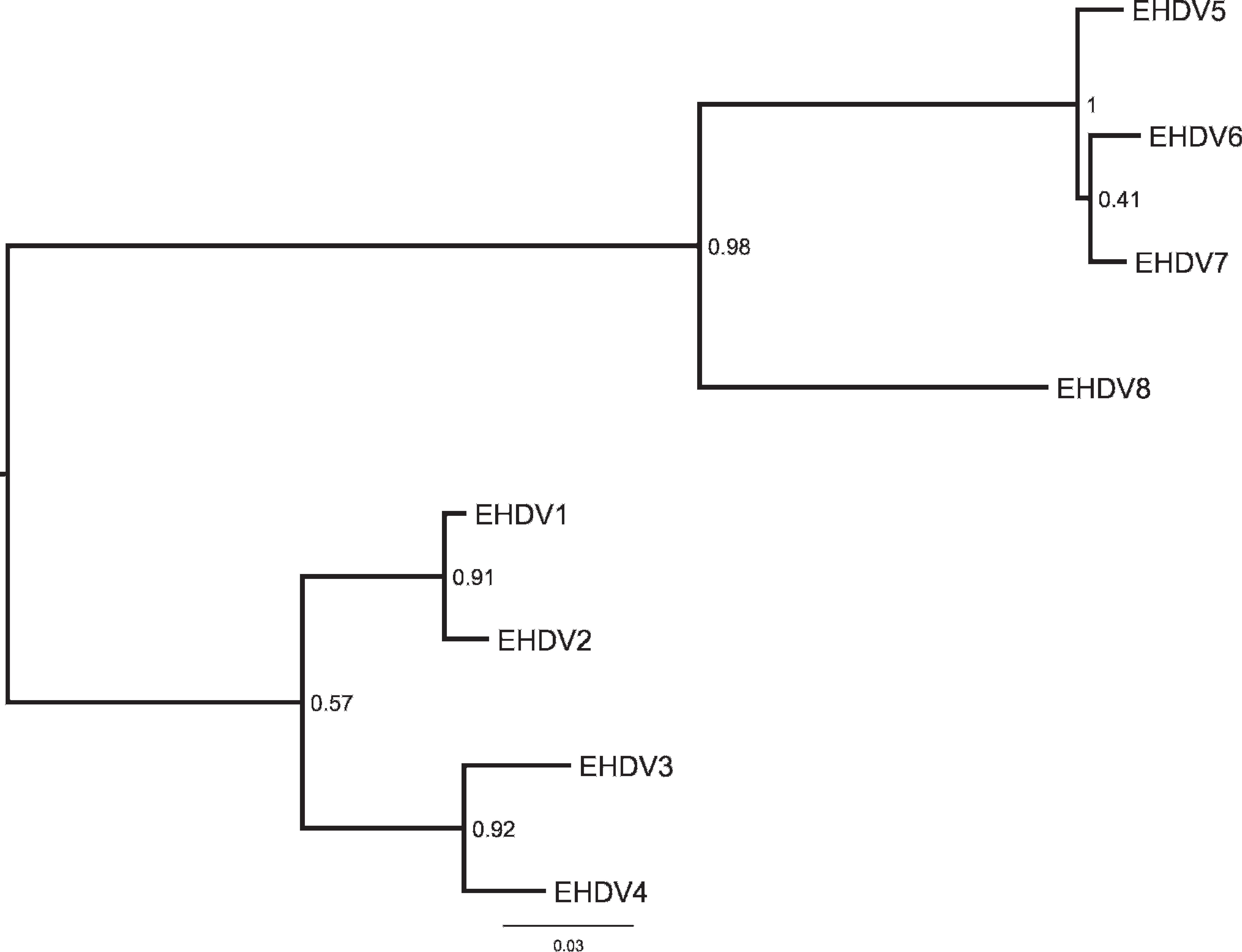
Phylogenetic analysis of the S10 gene from prototype Epizootic hemorrhagic disease virus (EHDV) serotypes. Sequences were aligned using MUSCLE, and the tree was calculated using MrBayes (750,000 generations, GRT+Γ+Inv). Numbers on nodes indicate posterior probabilities. Branch lengths are in nucleotide changes per site.
Surprisingly, after close examination of the M5 sequence alignment, conserved regions between all the serotypes of sufficient size to develop a real-time PCR primer set were not found. Therefore, the authors' attention turned to the second target for the primer design. Sequence analysis of the S10 gene from North American EHDV prototype and U.S. field strains showed 94.9–100% identity. 10,18 The S10 also had a greater diversity than expected; however, a region was identified to be suitably conserved to be used to design primers using Primer Express v. 2.0 software. c The primers were initially tested using SYBR Green RT-PCR c to verify amplification. Probes were labeled with a 5′ FAM reporter and 3′ BHQ-1 quencher. f For optimal detection of all 8 EHDV serotypes, the real-time RT-PCR assay used 7 primer sets and 3 probes. Probe 1/2 detected EHDV serotypes 1, 2, 5, 6, 7, and 8. Probe 3 detected EHDV serotype 3, and probe 4 detected EHDV serotype 4. Each serotype had its own amplification primer pair except serotypes 1 and 2, which shared a primer set (1/2; Table 1).
TaqMan EZ-RT PCR reagents c were used for the realtime RT-PCR in half-reaction volumes and included 0.4 μmol of each primer and 0.08 μmol of each probe. The cycling parameters were based on the previously reported real-time RT-PCR for Vesicular stomatitis virus. 30 The cycle temperatures were 55°C for 25 min, 95°C for 2 min, 40 cycles of 95°C for 10 sec, and 55°C for 1 min. The samples were analyzed using 3 real-time PCR instruments. c,g,h All 3 instruments performed similarly, and use was determined based on instrument availability and number of samples. Analysis of the limit of detection (LOD) was performed using dsRNA from all 8 serotypes of EHDV and was done in duplicates using 10-fold serial dilutions. The EHDV serotypes 1 and 2 using a single probe detected down to at least 2 fg (∼100 RNA copies). The assay using all 3 probes detected EHDV serotype 1 at 20 fg (∼1,000 RNA copies), serotypes 2 and 6 to at least 2 fg, serotypes 3 and 5 at 200 fg (∼10,000 RNA copies), and serotypes 7 and 8 at 2 pg (∼100 RNA copies). The sensitivity of the assay did not increase by the addition of a heat denaturant step. A 10-fold increase in sensitivity with methyl mercury hydroxide denaturation in the domestic BTV or EHDV real-time RT-PCR assays has been previously reported 32 ; however, this reagent is no longer commercially available. Other chemical denaturants were not as effective in improving sensitivity and reduced the amount of input template, and thus deemed not sufficiently advantageous to add a denaturation step.
Epizootic hemorrhagic disease virus (EHDV) real-time primer and probe sequences.
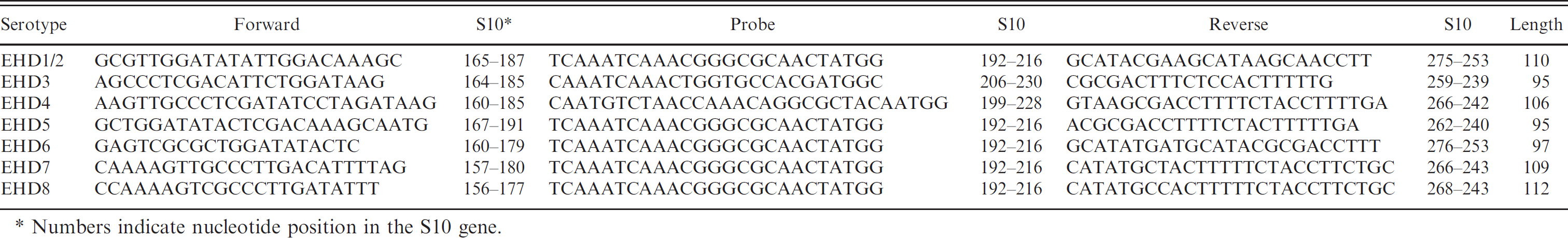
Numbers indicate nucleotide position in the S10 gene.
The LOD determination is only an estimate based on purified dsRNA input. To better evaluate the assay performance, the real-time assay was compared to the nested RT-PCR (nRT-PCR) 28 on archived clinical material. In addition, available samples were analyzed by virus isolation (VI) and were also confirmed by an antigen-capture ELISA. 12,16 Total RNAs from clinical samples were extracted using a commercial kit, b with minor modifications. The PCR assays were tested on 44 U.S. EHDV isolates, 36 tissue or blood samples from pronghorn antelope, white-tailed deer, and mule deer, and 23 archived clinical RNA samples, for 103 total samples. The U.S. isolates and clinical samples were mostly detected by the real-time RT-PCR assay at 200 pg of total RNA; however, 6 field strains and 8 clinical samples required a higher concentration. All 44 EHDV U.S. isolates tested positive by both real-time RT-PCR and nRT-PCR. Of the clinical samples, 19 tested positive by both real-time RT-PCR and nRT-PCR (Table 2). Additionally, this real-time RT-PCR assay was run against all 24 BTV serotypes to analyze cross-reactivity, and all were found to be negative.
Thirty-six of the archived clinical samples were tested by standard VI and antigen-capture ELISA to confirm the presence of EHDV. 16 Of VI-positive samples, 10 were positive by EHDV antigen-capture ELISA, for a prevalence of 28%. Two of the EHDV VI–negative samples were negative for EHDV by both nRT-PCR and real-time RT-PCR and were found positive by BTV antigen-capture ELISA. 12 One VI-positive sample contained both BTV and EHDV antigens as detected by the appropriate antigen-capture ELISAs. This sample was also positive for EHDV by both RT-PCR assays. Nine samples were VI negative but were positive for EHDV RNA by both RT-PCR assays. The real-time RT-PCR performed identically to the nRT-PCR with the 24 archived clinical RNA samples (Table 2).
The initial single-probe design only detected EHDV serotypes 1, 2, 5, 6, 7, and 8. Attempts to detect EHDV serotypes 3 and 4 using degenerative probes and modifying the protocol were unsuccessful. Additional probes were necessary for detection of these 2 serotypes. Thus, optimal detection of all 8 serotypes included the design of 7 amplification primers and 3 detection/confirmation probes. This EHDV all-serotype detection assay did not cross-react with any of the 24 BTV serotypes. This preliminary assay in the current study is based solely on the EHDV S10 target, but it performs identically to a previous EHDV nRT-PCR 28 that was designed solely for North American serotypes. This assay is an improvement over the previous assay in that it is a closed system (and thus has fewer cross-contamination problems), is less time consuming than traditional nRT-PCR, does not require a toxic methyl mercury denaturation step, and detects all 8 EHDV serotypes. Given the recent detection of exotic EHDV serotype 6 in the United States, this should prove to be a useful assay for veterinary diagnostic laboratories. Recently, increased sensitivity has been achieved using an alternative RNA extraction and detection method for use in a multiplex BTV/EHDV real-time RT-PCR assay (Wilson, Hindson, O'Hearn, et al., unpublished data). Future improvements will include additional primer sets targeting the M5 gene to provide further assay assurance and also to multiplex using a similar assay currently in development for simultaneous BTV detection.
Epizootic hemorrhagic disease virus (EHDV) real-time reverse transcription polymerase chain reaction (real-time RT-PCR) compared with EHDV nested RT-PCR and EHDV virus isolation (VI) on various samples.*
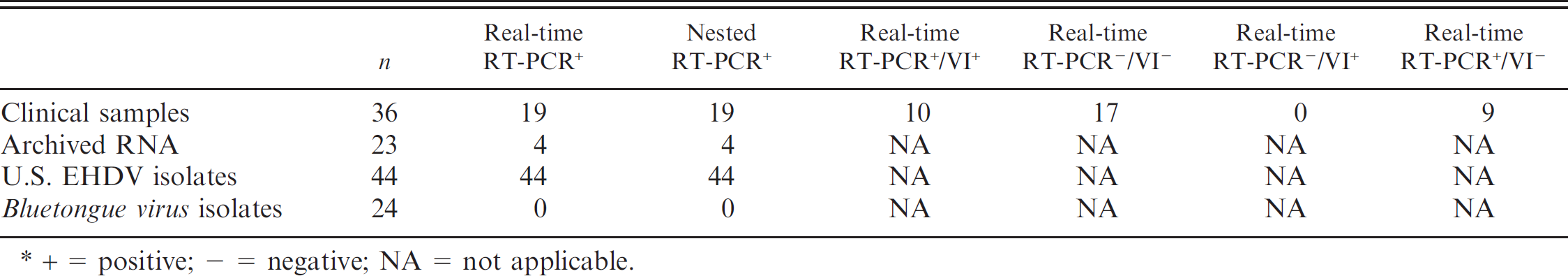
+ = positive; – = negative; NA = not applicable.
Acknowledgements. The authors acknowledge Donna Johnson and Dr. Eileen Ostlund from Animal and Plant Health Inspection Service-National Veterinary Services Laboratories (Ames, IA) for providing RNA from the exotic strains of BTV and EHDV necessary to complete this study. This project was funded by U.S. Department of Agriculture-Agricultural Research Service projects 5410–32000–011–00D and 5410–32000–016–00D. Bioinformatic analysis was supported by National Institutes of Health grant P20 RR016474 from the IDeA Networks of Biomedical Research Excellence Program of the National Center for Research Resources. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH or the USDA.
Footnotes
a.
Purescript® RNA Isolation Kit, Gentra Systems Inc., Minneapolis, MN.
b.
Qiagen RNeasy Kit, Qiagen Inc., Valencia, CA.
c.
Invitrogen Inc., Carlsbad, CA.
d.
Applied BioSystems Inc., Foster City, CA.
e.
DNA Star Inc., Madison, WI.
f.
Biosearch Technologies Inc., Novato, CA.
g.
Bio-Rad Laboratories, Hercules, CA.
h.
Cepheid Inc., Sunnyvale, CA.