
Abstract
Progressive rod-cone degeneration (prcd) is a late onset, autosomal recessive, inherited disease in dogs caused by a G > A substitution in the PRCD locus, which has been reported in more than 18 breeds, including Labrador Retriever dogs. In this study, a real-time polymerase chain reaction (PCR) assay, exploiting the features of locked nucleic acid (LNA) fluorescent-labeled probes, was developed to genotype the sequence variants responsible for the disease. Two Labrador Retrievers were diagnosed with prcd by ophthalmological examination performed by a panelist of the Italian hereditary eye disease control program. The 2 dogs, as well as 8 related and 14 unrelated Labrador Retrievers, were genotyped with both direct sequencing of the disease locus and real-time LNA TaqMan PCR assay. Even though the region surrounding the mutation was predicted to be highly structured, making probe annealing difficult, the real-time PCR assay allowed researchers to correctly genotype the dogs in all cases with a sensitivity threshold of 4 ng/reaction of genomic DNA. A real-time PCR assay will allow a high-throughput analysis of a larger cohort of dogs, thereby enabling researchers to investigate the prevalence of the mutated allele in the affected breeds.
Keywords
Progressive retinal atrophies (PRAs) are a group of heterogeneous inherited retinal disorders in dogs that share many phenotypic and clinical features with human retinitis pigmentosa. PRAs cause a progressive visual deterioration, usually leading to blindness. 5 Early clinical signs are represented by “night blindness,” mydriasis, and increased eye reflectivity noticed by the owners. 5 The disease usually worsens, leading to daytime blindness. On the basis of the age of onset and the rate of progression, PRAs can be classified as early- or late-onset forms. Diagnosis is based on striking clinical signs, ophthalmologic examination of the fundus, and electroretinography. 5 Regardless of the phenotypic heterogeneity, the known mutations responsible for PRAs occur in many different genes typically involved in the phototransduction pathways. One of the more common forms of PRAs, known as progressive rod-cone degeneration (prcd), is a late-onset, autosomal recessive disease reported in 18 breeds to date. 6 In all 18 breeds tested, prcd is caused by a c.5G > A (DQ390330) allelic variant of a gene of unknown function called PRCD, which codes a 54-amino acid protein and maps on a region of canine chromosome 9 syntenic with human chromosome 17. 6 Molecular tools that could be used effectively to genotype known single-nucleotide polymorphisms (SNPs) are numerous. Along with the increasing demand of facile and robust SNP typing, new methods for carrying out allelic discrimination by real-time polymerase chain reaction (PCR) assays have been proposed in recent years. Real-time PCR with dual-labeled probes modified with locked nucleic acids (LNAs) has been successfully used to genotype the SNP responsible for polycystic kidney disease in Persian cats. 2 This method relies on the simultaneous use of 2 internal hybridizing probes that perfectly match the sequence of the 2 alleles. The SNP residue on the target DNA corresponds to an LNA base in both probes. LNAs are nucleic acid analogues that are conformationally locked in a C3-endo/N-type sugar conformation by an O-2’ to C-4’ methylene linkage. 1,4 The LNA/DNA duplex shows much increased stability and a higher melting temperature. Thus, the greater difference between matched hybrids and less stable mismatched hybrids allows allelic-specific hybridization and enhances allelic discrimination. 3
The LNA-modified probes carry a different fluorogenic molecule at the 5’ end and a quencher molecule at the 3’ end. On primer extension, Taq polymerase nuclease activity cleaves the allele-specific probe with a simultaneous increase of the respective fluorescence signal. In this study, the authors report the setup of a rapid and accurate realtime PCR assay to genotype the allelic variant responsible for prcd in Labrador Retrievers.
Two Labrador Retrievers affected by prcd, 8 dogs related to those affected, and 14 unrelated Labrador Retrievers were included in the study. Each dog underwent clinical examination, including fundus ophthalmologic assessment. The dogs were 1–12 yr of age, and there were 11 males and 13 females. Each dog was genotyped by direct forward and reverse sequencing. Genomic DNA (gDNA) was extracted from anticoagulated K3-ethylenediamine tetra-acetic acid blood samples with a DNA extraction kit a following the manufacturer's instructions. A primer pair (forward: 5‘-AGCCTCCTAATCCAGTGG-3′; reverse: 5‘-GTGCTCT GATGGGAAACC-3′) was designed with commercial software b to amplify a 346-bp fragment of gDNA encompassing the disease locus. The PCR products of each sample were directly sequenced from both strands with the use of a commercial sequencing kit c on a genetic analyzer. c The sequences were analyzed with commercial software. c
The complete PCR sequence was used to design the real-time PCR assay with dual-labeled, LNA-modified TaqMan probes. The SNP region was predicted to have poor quality for probe binding because of heavily folded sequences, even at 60°C. The design of 2 different real-time PCR assays employing different primer/probe sets was realized by Proligo d on both the positive and negative strands. The LNA probes designed on the positive strand (PS) should discriminate a G > A substitution (PS assay), whereas the LNA probes designed on the alternate strand (NS) should discriminate a C > T substitution (NS assay). The expected PCR product length of the PS and NS assays was 124 and 249 bp, respectively. Because of the target sequence features (highly self-complementary), the probes were designed with a limited number of LNA modifications to avoid the formation of primer dimers. The PS and NS wild-type probes were labeled with a 5’ hexachloro4fluorescein (HEX) moiety and a black hole quencher 1 (BHQ1) moiety at the 3’ end, whereas the PS and NS prcd probes were dual-labeled with a fluorophore 6-carboxyfluorescin (FAM) moiety at the 5’ and a 3’ BHQ1. Melting temperatures were predicted with the web-based LNA melting temperature (Tm) calculator e and prediction conditions of 115 mmol salt concentration and 2 μmol of total oligo concentration. The predicted Tm were as follows (LNA residues are indicated by a + sign in front of the modified base): PS forward primer (5‘-GCAGACTCTGTCCGGGAG-3′) 65°C, PS reverse primer (5‘-ATCAGCTTCTCACGGTTGGA-3′) 63°C, PS wild-type LNA probe (5‘-[HEX]AGCCATGT[+G]C[+A]CCACCCT[BHQ1]-3′) 71°C, PS prcd LNA probe (5‘-[6FAM]AGC[+C]ATGT[+A]C[+A]CCACCCT[BHQ1]-3′) 70°C, NS forward primer (5‘- AGCCTCCTAATCCAGTGG-3‘) 61°C, NS reverse primer (5′-CCTAGCTTCTCCTCTCTCC-3′) 61°C, NS wild-type LNA probe (5′-[HEX]AGGGT[+G]GTG[+C]ACATGGCTC[BHQ1]-3′) 75°C, and NS prcd LNA probe (5′-[6FAM]AGGGT-[+G]GTG[+T]ACAT[+G]GCTC[BHQ1]-3′) 75°C. The secondary structure of all primers and probes of the PS and NS assays were checked separately with the LNA probe web-based optimizer software. e
A real-time genotyping assay was carried out in a thermal cycler f with the use of 12.5 μl of 2x Master Mix, f different concentrations of primers and probes, and 2 μl of gDNA (∼20–80 ng) brought up to 25 μl with molecular biology-grade water. The optimal primer concentrations were established with the use of different combinations of forward and reverse primer, which resulted in the highest ΔRn (normalized reporter fluorescence value) and the lowest threshold cycle (Ct). In the primer matrix experiment, different concentrations of 100, 300, 600, and 900 nmol for each primer were tested in triplicate (i.e., 100/100, 100/300, 100/600, 100/900, 300/100, 300/ 300, 300/ 600, 300/900, 600/100, 600/300, 600/600, 600/900, 900/600, and 900/900). The optimal primer concentrations were used with different probe concentrations according to a matrix encompassing 50, 75, 100, 150, 200, 250, and 400 nmol with the use of both wild-type and mutated homozygous templates, as well as a heterozygous template. Again, the concentration of primers and probes yielding the highest ΔCt between the 2 fluorophore probes in both homozygous wild-type and mutated samples, as well as a ΔCt within 3Ct in the heterozygous template, were selected. Finally, 10-fold serial dilutions of gDNA of homozygous and heterozygous samples were tested to assess sensitivity. The PCR protocol included an initial denaturation at 94°C for 2 min followed by 45 cycles of 94°C for 15 sec, annealing at 55°C for 20 sec, and extension at 68°C for 20 sec when fluorescence acquisition was accomplished.
The FAM was read at 520 nm, whereas the HEX fluorescence was collected by using the VIC® fluorophore channel at 550 nm. Data was acquired in real time to calculate the Ct values and further postprocessed as an endpoint to calculate the fluorescence. Results were analyzed by the discriminant analysis option of a statistical software program. g
Although both sets of primers efficiently amplified the target (results not shown), the primers and probes for NS genotyping did not yield any signal. On the contrary, the oligos designed for genotyping on the positive strand performed well with a 900-nmol forward and reverse primer concentration and an unbalanced probe concentration of 250 and 50 nmol of HEX- and FAM-labeled probes, respectively. Both the Ct and ΔRn methods could readily distinguish among heterozygous, wild-type homozygous, and mutant homozygous genotypes (Fig. 1). The discriminant analysis correctly assigned all subjects to the 3 genotypes with an associated probability of >99.9%.
A 10-fold serial dilution of gDNA samples showed that the assay yielded reliable results with as little as 4 ng/ reaction of gDNA. Below that threshold, inconsistent results were found.
A rapid and accurate method of genotyping the mutation responsible for prcd in Labrador Retrievers is herein presented. Although the LNA probe that was based on the positive strand sequence worked well, the probe that was based on the negative strand sequence did not. This is not surprising because oligonucleotide interactions when LNA bases are added are somewhat unpredictable and stresses out the difficulties in designing LNA probes.
This method allowed researchers to obtain results in approximately 2 hr compared with the longer times needed by alternative techniques, such as restriction length fragment polymorphism and direct sequencing. Furthermore, real-time PCR allelic discrimination assays do not involve the opening of reaction tubes, thus avoiding the possibility of carryover and false-positive results.
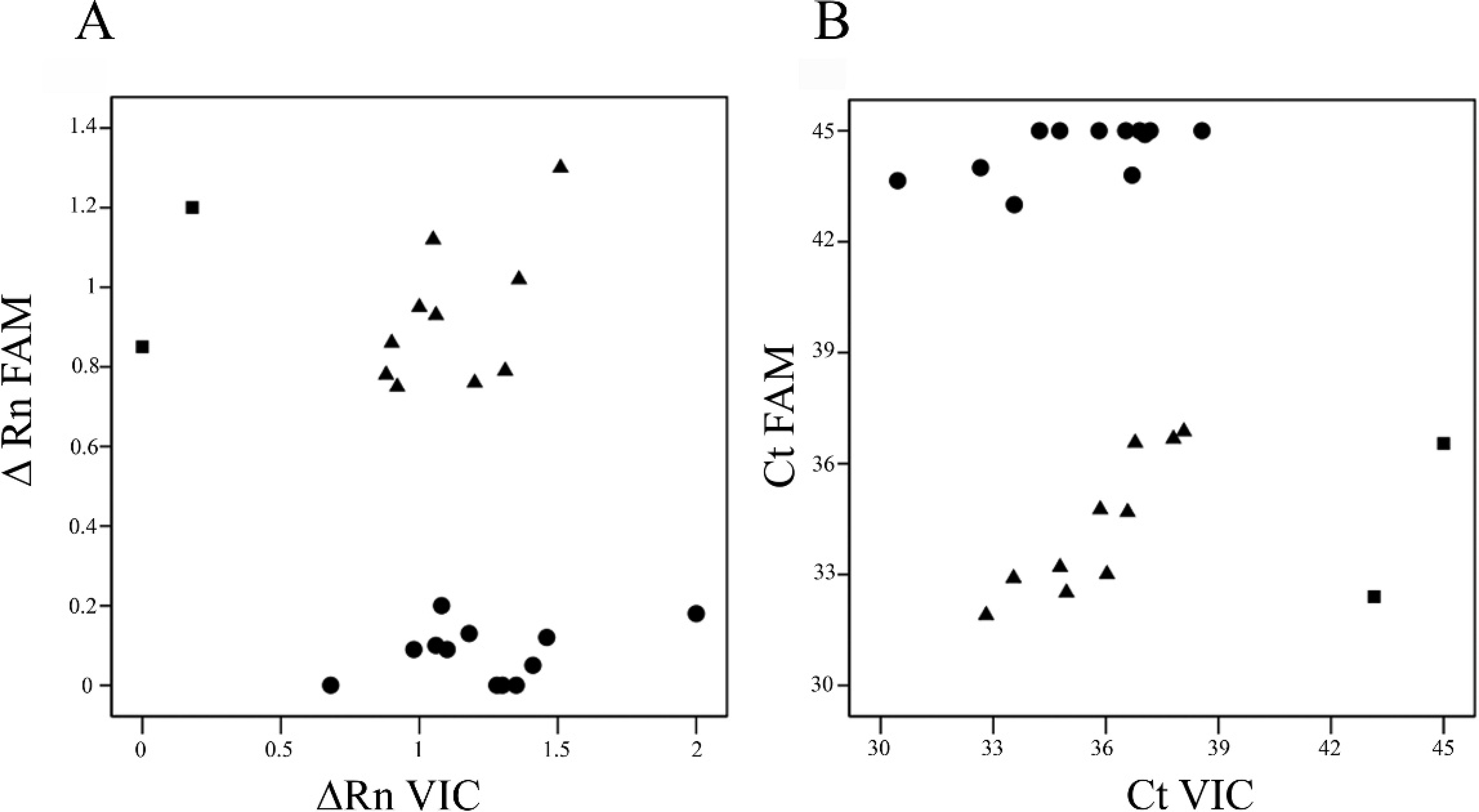
Real-time polymerase chain reaction results of an allelic discrimination assay.
Expensive equipment, other than that commonly used for real-time PCR, is not required. The LNA modified probes are as expensive as other fluorescent-labeled probes. Although the overall cost for each assay is higher than the cost of other high-throughput sequencing methods such as pyrosequencing, pyrosequencing requires highly expensive equipment.
The assay was accurate, with a sensitivity shown to reach approximately 4 ng/reaction of gDNA. Limited sensitivity is likely due to the above-mentioned target sequence features (highly self-complementary). Indeed, extensive folding of both the SNP flanking region and the overall amplicon are predicted to occur even at 60°C. However, such a sensitivity threshold is not a great concern because a gDNA yield of more than 4 ng is commonly obtained from biological samples used for genetic testing, including anticoagulated blood and buccal swabs.
Although genetic testing for prcd, both initially performed with a linkage test and, in later years, with mutation detection assays, has been available for almost a decade, the causative allelic variant prevalence is unknown. Realtime PCR is particularly suitable for high-throughput screening of large samples for retrospective studies aimed at assessing mutation prevalence. Knowledge of the actual diffusion of the allelic variant might help in improving awareness in breeders of the necessity of introducing genetic testing into breeding programs involving Labrador Retrievers.
Acknowledgements. M. E. Turba conducted the study as a fulfillment of her Ph.D. research project in “Applicazioni Biotecnologiche in Neuromorfofisiologia-XIX ciclo” at the Department of Veterinary Morphophysiology and Animal Production, DIMORFIPA, Alma Mater Studiorum-University of Bologna, Italy.
Footnotes
a.
NucleoSpin® Blood, Macherey-Nagel GmbH & Co. KG, Düren, Germany.
b.
Beacon DesignerTM version 3, PREMIER Biosoft International, Palo Alto, CA.
c.
BigDyeTM Terminator v1.1, ABI PRISMTM 310 Genetic Analyzer, SeqScape® v2.5; Applied Biosystems, Milan, Italy.
d.
Sigma-Aldrich Srl, Milan, Italy.
e.
Exiqon, Vedbaek, Denmark.
f.
Real-Plex Ep-gradient S, Real Master Mix Probe; Eppendorf, Milan, Italy.
g.
SPSS v13.0 for Windows, SPSS Italia, Bologna, Italy.