
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is an important pathogen of swine. The objective of the current study is to investigate the feasibility of using reverse transcription loop-mediated isothermal amplification (RT-LAMP) for the detection of PRRSV. The RT-LAMP is a recently described DNA amplification technique reported to be simple, inexpensive, fast, and accurate. The RT-LAMP reaction was set up using 2 sets of primers that were designed to detect North American and European strains of PRRSV and performed successfully in a simple heat block. The specificity of the amplified product was demonstrated by restriction analysis. The RT-LAMP was able to detect 5 different PRRSV isolates. However, the limit of detection ranged between 10 2 and 10 4 50% tissue culture infective dose/ml. The RT-LAMP was further evaluated using serum samples from animals of known infection status. The ability of RT-LAMP to detect PRRSV in serum from acutely infected animals was evaluated with 114 serum samples from 18 experimentally inoculated boars. Forty-nine of these samples tested positive by RT-LAMP, while 94 were positive by reverse transcription polymerase chain reaction (RT-PCR). The diagnostic specificity, evaluated with 100 known negative serum samples, was estimated as 99%. The feasibility of RT-LAMP to detect PRRSV was demonstrated in the current study. The RT-LAMP reaction could be performed in just 1 hr with a simple and inexpensive heat block. However, the sensitivity of this technique was significantly lower than that of RT-PCR.
Porcine reproductive and respiratory syndrome is an important disease of swine caused by Porcine reproductive and respiratory syndrome virus (PRRSV), a RNA virus from the order Nidovirales, genus Arterivirus, and family Arteriviridae. This virus, which causes abortions in pregnant sows and respiratory disease in growing pigs, has a tremendous economic impact in swine production worldwide. 16 Reverse transcription polymerase chain reaction (RT-PCR), a highly sensitive and specific test, is considered the best option for PRRSV detection during the acute phase of infection. 13,15 Unfortunately, RT-PCR requires sophisticated equipment, specialized labor, and cannot be implemented successfully outside a specialized laboratory.
A novel diagnostic technique called loop-mediated isothermal amplification (LAMP) has been described. 11 Like PCR, this technique uses DNA primers and a polymerase enzyme to amplify a specific fragment of DNA of the target pathogen. However, because the LAMP reaction occurs at a constant temperature of 63°C, it can be performed in a simple water bath or heat block. The sensitivity and specificity of LAMP have been reported to be comparable with those of PCR. 4,11,14 Coupled with reverse transcription (RT-LAMP), the LAMP technology has been used to detect RNA viruses such as Severe acute respiratory syndrome coronavirus, 4 Human influenza virus, 12 and Foot-and-mouth disease virus. 3 The objective of the current study was to investigate the feasibility of using RT-LAMP for the detection of PRRSV.
A conserved stretch of at least 200 base pairs (bp) is needed to design LAMP primers. Because of the large divergence between the North American and European strains of PRRSV, a duplex RT-LAMP test including a set of primers for each strain was attempted. The Web-based primer design software Primer Explorer V4 a was used to design the primers targeting the conserved regions as recommended by the software developer. Four primers (2 inner primers and 2 outer primers) that hybridize with 6 specific sequences of the target DNA are used in LAMP (Table 1). 11 In addition, 2 more primers (loop primers) can be used to accelerate the reaction. 10
RNA was extracted from virus culture and serum samples using a commercial kit b following the manufacturer's instructions. The RT-LAMP reaction was performed in a 25-μl reaction mixture containing 1.6 μmol of each inner primer, 0.2 μmol of each outer primer, 0.8 μmol of each loop primer, 1.4 mmol of each deoxyribonucleotide triphos-phate, c 0.8 mol of betaine, d 2.5 μl of 10× Thermo buffer, e 8 mmol MgSO4, e 8 U of Bacillus stearothermophilus DNA polymerase, e 2 U of enhanced Avian myeloblastosis virus reverse transcriptase, d and 2 μl of the extracted target RNA. 4,11,14 The reaction was performed in a heat block f at 63°C for 1 hr. The detection of the RT-LAMP reaction product was performed by electrophoresis in a 2% agarose gel stained with ethidium bromide. The RT-LAMP reaction product is a combination of fragments of DNA of different sizes. Therefore, the presence of a smear or a pattern of multiple bands of different molecular weights was considered a positive result. 11 A molecular marker g was used to estimate product size.
DNA sequences of the primers used in the reverse transcription loop-mediated isothermal amplification in the current study.

A restriction analysis was performed on the RT-LAMP product to confirm the specificity of the reaction. The exact nucleotide sequence of the RT-LAMP products can be predicted from the sequences of the target DNA and the primers. Therefore, it is possible to predict the outcome of the digestion of the sample with a restriction enzyme that cuts the DNA in specific recognition sites. This method serves as a control of the specificity of the product of the LAMP reaction because the unspecific amplification of DNA would yield a different pattern of bands after digestion. 3,4,11 Restriction enzymes were selected with the NEBcutter version 2.0 online software. e For the restriction analysis of the PRRSV North American strain product, the PRRSV isolate MN30-100 and the enzyme EaeI e were used. For the restriction analysis of the product from the European PRRSV strain, the Lelystad virus isolate and the restriction enzyme NlaIV e were used. Digestion reactions were performed by incubating overnight 2 μl of RT-LAMP product obtained from a sample containing 10 5 50% tissue culture infective dose (TCID50)/ml of PRRSV, in the presence of 12 U of restriction enzyme in a total volume of 25 μl. The size of the digested fragments was estimated by agarose gel electrophoresis.
In addition to the detection of the amplified product by electrophoresis in agarose gel as described above, 3 alternative detection methods were evaluated: 1) Turbidity: the accumulation of magnesium pyrophosphate, a by-product of the DNA amplification reaction, results in the increase of the turbidity of the sample. 9 The turbidity was evaluated by visual inspection of the samples, comparing them to a negative control. 2) Color change: 1 ml of 10,000× SYBR Green I nucleic acid stain h was added to the tube after the reaction. Samples turning yellow-green were considered positive, while samples turning orange were considered negative. 14 3) Fluorescence from calcein: calcein is a chelating molecule that binds to manganese. If manganese is released, the calcein molecule becomes fluorescent. In a positive RT-LAMP reaction, the pyrophosphate binds to the manganese ion, releasing free calcein, which emits fluorescence. 1 To evaluate this method of detection, 1 μl of fluorescent reagent i containing calcein was added to the LAMP reaction mixture before the reaction. Samples showing fluorescence under an ultraviolet (UV) hand lamp j at a 365-nm wavelength were considered positive. Samples were compared with a negative control to account for background fluorescence. To compare the methods of turbidity, color change, fluorescence from calcein, and agarose gel electrophoresis, an RT-LAMP reaction was performed on replicates of a sample containing 10 4 TCID50/ml of PRRSV North American strain MN30–100. To generate samples with different amounts of RT-LAMP product, the replicates were allowed different reaction times of 20, 30, 40, 50, and 60 min. Each replicate was matched with a negative control sample, and all replicates were run simultaneously.
To evaluate the analytic sensitivity, 5 well-characterized PRRSV isolates and 2 attenuated isolates used in modified live vaccines were grown in cloned monkey kidney (MARC 145) cells. Six of the isolates (VR2332, MN30–100, MN-184, JA-142, ATP, and MLV) were North American-type isolates, and 1 (Lelystad virus) was a European-type isolate. Each isolate was titrated and diluted in culture media to obtain 10-fold dilutions containing 10 5 to 10−1 TCID50/ml. RNA extraction and RT-LAMP was performed on each dilution as described above to determine the limit of detection for each isolate. To evaluate its analytical specificity, the RT-LAMP test was performed on a panel of viral isolates commonly isolated from pigs. The panel included Influenza virus H1N1 and H3N2, Porcine respiratory coronavirus, Porcine pseudorabies virus, Encephalomyocarditis virus, Porcine enterovirus, Bovine viral diarrhea virus 1 and 2, Hepatitis E virus, Porcine parvovirus, Porcine cytomegalovirus, and Porcine circovirus 1 and 2.
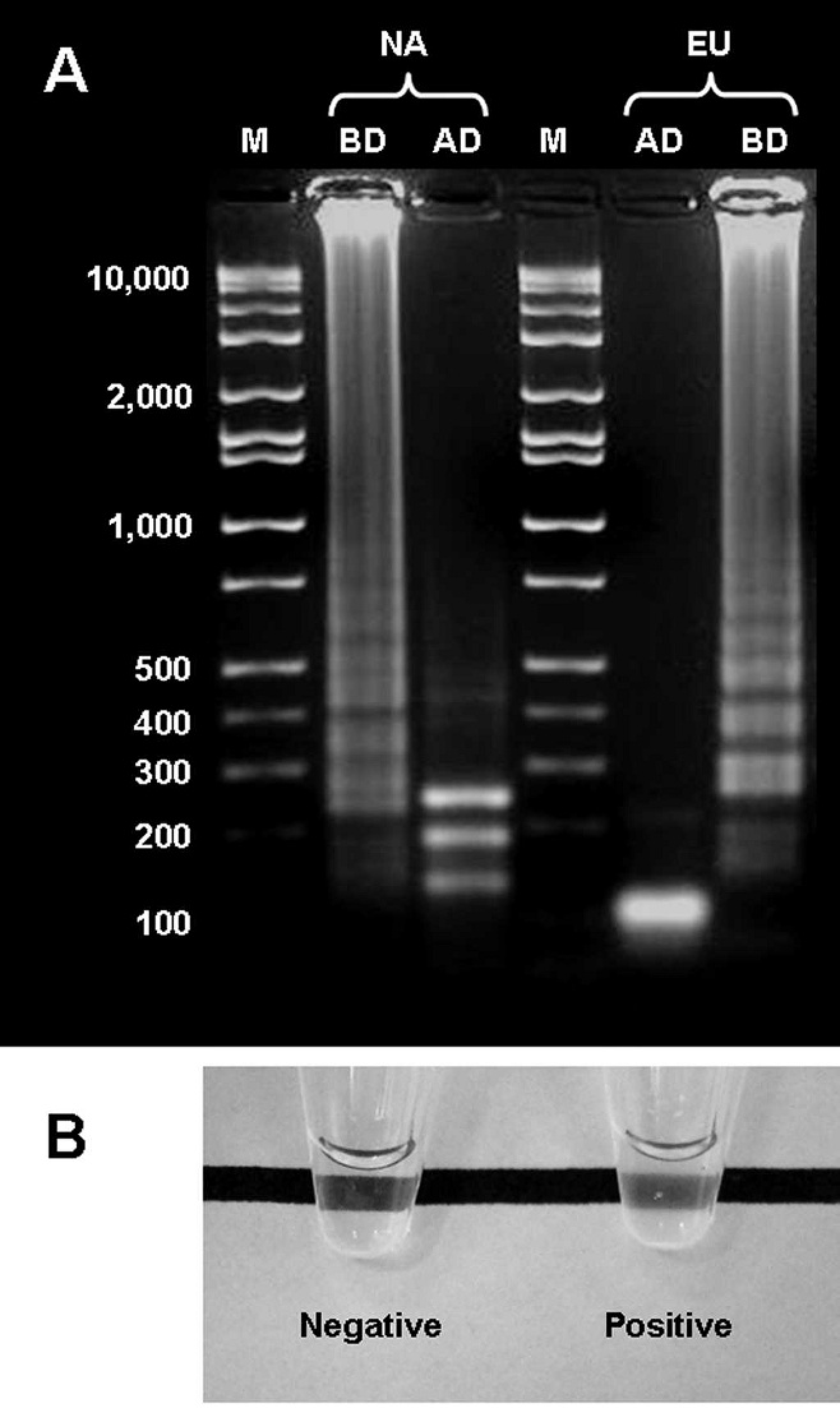
Detection of the Porcine reproductive and respiratory syndrome virus (PRRSV) reverse transcription loop-mediated isothermal amplification (RT-LAMP) products. A, agarose gel electrophoresis visualization of the RT-LAMP products from a PRRSV North American strain (NA) and a PRRSV European strain (EU), before (BD) and after digestion (AD) with restriction enzymes. The sizes of the main bands of the molecular marker (M) are indicated in base pairs. For a better visualization of the bands, 5 times less product was loaded in AD compared with BD. B, detection of RT-LAMP products by turbidity. Positive and negative control samples are shown.
To evaluate the ability of RT-LAMP to detect PRRSV in serum samples from acutely infected animals, the test was performed on 114 serum samples from 18 experimentally inoculated boars that were part of a previous study. 13 These samples were obtained at 1, 3, 5, 8, 10, 12, and 15 days postinoculation with PRRSV, and 94 of them tested positive for PRRSV by RT-PCR. 13 Diagnostic specificity (proportion of truly negative samples that test negative by RT-LAMP) was estimated using serum samples from 100 pigs from known PRRSV-negative farms.
Thirty-two North American PRRSV whole-genome sequences available in GenBank were aligned. 8 A relatively conserved region of 330 bp was found between nucleotides 14,591 and 14,920 of the reference VR-2332 strain (GenBank accession no. U87392). Similarly, 7 whole-genome RNA sequences of the European PRRSV strain available at GenBank were aligned. A relatively conserved region was identified between nucleotides 14,769 and 15,134 of the reference Lelystad virus strain (GenBank accession no. A26843), and primers targeting this region were designed (Table 1). A basic local alignment search tool (BLAST) k analysis for each target sequence in the designed primers showed 100% identity only with previously published sequences of PRRSV.
The RNA was successfully reverse transcribed, and the DNA was amplified from both North American and European strains of PRRSV by RT-LAMP (Fig. 1A). Negative control (culture media) and nontemplate samples were negative. Taking into account the target sequence, the restriction sites, and the different possibilities for the theoretical arrangement of the amplified products, 11 the digestion of the RT-LAMP products from the North American PRRSV strain should result in 3 main fragments of 242, 187, and 132 bp. Figure 1A shows the RT-LAMP product before and after digestion with EaeI. Before digestion, the product looks like a smear because of the presence of DNA fragments of multiple sizes ranging from more than 10,000 to less than 200 bp. After digestion, 3 bands are distinguishable, 1 of size between 300 and 200 bp and 2 of sizes between 200 and 100 bp. The 3 bands are consistent with the predicted sizes of 242, 187, and 132 bp. Similarly, the digestion of the RT-LAMP products from the European PRRSV strain should result in 2 main fragments of 101 and 113 bp. Figure 1A shows that after digestion, there was a single, very intense band of about 100 bp, consistent with the predicted fragment sizes.
The products of the RT-LAMP reaction were detected by agarose gel electrophoresis after 40, 50, and 60 min of reaction but not after only 20 or 30 min. With the detection methods of turbidity (Fig. 1B) and change in color, only the samples run for 50 and 60 min were positive. Positive and negative samples were easily distinguishable with these 2 methods. In the samples in which calcein was added, background fluorescence was observed under UV light in the negative control samples. Samples run for 20 and 30 min showed a weak fluorescent signal comparable with that of the negative control. The samples run for 50 and 60 min showed a strong fluorescent signal, more intense than the background fluorescence. More difficult was the interpretation of the sample run for 40 min, which showed an intermediate fluorescent signal. Based on these results, the evaluation of the analytic sensitivity and specificity of RT-LAMP and further evaluation with serum samples were performed using the turbidity and change in color methods only.
The limit of detection was 10 2 TCID50/ml for isolates JA-142 and Lelystad virus; 10 3 TCID50/ml for isolates MN30-100, MN-184, and MLV; and 10 4 TCID50/ml for isolates ATP and VR2332. The RT-LAMP test was negative for all the samples in the panel of viruses frequently isolated from swine. Of the 114 samples from experimentally infected boars, 49 tested positive for PRRSV by RT-LAMP. Therefore, RT-LAMP detected PRRSV in 43% of all the serum samples and in 52% of the PCR-positive samples. Of the 100 known negative serum samples, 99 tested negative by RT-LAMP. This represents a diagnostic specificity of 99%. The only sample that tested positive was retested in duplicate, with negative results.
The feasibility of RT-LAMP to detect PRRSV was demonstrated in the current study. With this new diagnostic technique, it is possible to detect PRRSV by specifically amplifying a DNA fragment complementary to the RNA of the virus and detecting the amplified product. The RT-LAMP reaction can be performed in just 1 hr with a simple and inexpensive heat block.
Four different detection methods were evaluated. The electrophoresis in agarose gel is a very sensitive method to detect DNA and was used as a reference method. This method was slightly more sensitive than the other 3 methods. However, this method is not intended for routine use because it is time-consuming and requires specialized equipment. Under the conditions of the present study, the detection method based on fluorescence from calcein could be subject to misinterpretation because of the presence of fluorescent background on negative samples. The methods of turbidity and change in color were selected for their simplicity and ease of interpretation. These 2 methods were further compared with the serum samples used for validation and yielded identical results in each of the 214 serum samples tested. However, the method of change in color requires opening the tube at the end of the reaction. This represents an important limitation for an on-site test because of the high chances of contamination with the end product of the reaction. For that reason and because of its simplicity, the detection based on turbidity is preferred. The limitation of this method is that the interpretation can be somewhat subjective. To address this limitation, other studies used a special heat block that has a turbidimeter incorporated. 4 This device can measure the turbidity of the sample with high accuracy in real time. Nevertheless, in the present study, positive and negative samples were clearly distinguishable, and the results with this method perfectly matched those obtained with the change in color method.
Since 6 different sequences on the target DNA are recognized by the LAMP primers, this is theoretically a very specific technique. 11 In the present study, the restriction analyses confirmed the specificity of the amplified DNA fragments. The evaluation of the specificity was completed by the lack of amplification of DNA from other swine viruses and by testing 100 known negative serum samples. One of these, 100 samples tested positive. Since this sample tested negative after retest, the false-positive result is likely due to contamination of the tube while setting up the reaction.
The main challenge in the development of molecular tests for PRRSV is the large genetic heterogeneity of this virus. 5,7 In the current study, 5 different PRRSV isolates and 2 attenuated variants were detected by RT-LAMP. After the development of the test, 10 additional field isolates (9 of the North American strains and 1 of the European strains) were tested, and 9 of them were successfully detected by RT-LAMP (data not shown). The isolate that could not be detected was a North American variant (known as 1-18-2) that recently spread in the Northwest United States. This emphasizes the need for a constant update of the primers of molecular diagnostic tests for PRRSV. Most current RT-PCR tests include 2 or more sets of primers to capture most of the variability observed in the PRRSV genome. 7 Multiplex tests have been described for the LAMP technique as well. 2,6 In the current study, 2 sets of primers were included, and it is likely that a third set of primers could be added without a detrimental effect on the performance of the test.
The main limitation of the diagnostic test developed was the sensitivity. The limit of detection ranged from 102 to 104 TCID50/ml, far from the limits reported for RT-PCR tests. 7,15 The results obtained from serum samples also showed a reduced sensitivity compared with RT-PCR. Specifically, the RT-LAMP failed to detect the virus in samples with low copy numbers of virus (data not shown), mainly obtained during the first 1 to 5 days postinoculation. 13
To the authors' knowledge, this is the first study that has explored the use of RT-LAMP technology in a diagnostic test for PRRSV. The test was simple, specific, and rapid and was able to detect PRRSV from serum samples from acutely infected animals. However, the sensitivity was lower than that of RT-PCR. Nevertheless, there is potential for this technique to be applied in situations in which RT-PCR is too expensive or too sophisticated to be implemented.
Acknowledgements. Dr. Goyal (University of Minnesota) kindly provided the panel of viral isolates. The PCR work was performed by Dr. Christopher-Hennings and her research team (South Dakota State University). This project was funded by the National Pork Board (project 06–154).
Footnotes
b.
QIAamp® Viral RNA Mini-Kit, Qiagen Inc., Valencia, CA.
c.
Master Mix dNTP, Denville Scientific Inc., Metuchen, NJ.
d.
Sigma-Aldrich, St. Louis, MO.
e.
New England Biolabs Inc., Ipswich, MA.
f.
D1200, Labnet International Inc., Woodbridge, NJ.
g.
Hi-Lo DNA Marker, Minnesota Molecular Inc., Minneapolis, MN.
h.
Molecular Probes Inc., Eugene, OR.
i.
Loopamp®, Eiken Chemical Co., Tokyo, Japan.
j.
Spectroline® Enf-24, Spectronics Corp., Westbury, NY.
k.
BLAST, National Center for Biotechnology Information, Bethesda, MD.