
Abstract
Sheeppox and goatpox are economically important viral diseases of sheep and goats, respectively. Both diseases are reportable to the World Organization for Animal Health. To implement a control and eradication program for these diseases, a rapid and user-friendly diagnostic tool is imperative for screening. Therefore, in the present study, TaqMan quantitative polymerase chain reaction (qPCR) and conventional PCR assays targeting the DNA polymerase (DNA pol) gene were developed for the detection of Capripoxvirus DNA from clinical specimens of sheep and goats. The 2 assays used different primer sets. Conventional PCR yielded a specific product of 134 bp, whereas qPCR yielded a 180-bp product. The specificity of amplified DNA pol gene products was confirmed by their size and by sequence analysis. The 2 assays were specific for Sheeppox virus and Goatpox virus. However, in comparison to conventional PCR, the qPCR was more rapid, specific, and 100 times more sensitive, with a detection limit as low as 0.042 pg of purified DNA. The qPCR assay was more sensitive (84.05%) than conventional PCR (76.06%) when used on clinical samples (n = 71) from sheep and goats.
Small ruminants comprise a vital component of livestock farming and provide livelihood to millions in the farming sector. The world population of sheep and goats is approximately 2.1 billion. 4 Sheeppox and goatpox (collectively known as capripox diseases) are the most important World Organization for Animal Health notifiable viral diseases that pose serious socioeconomic impact to small-ruminant productivity in terms of morbidity, mortality, hide damage, and trade restrictions. Countries that are free from capripox diseases enjoy a significant advantage in the export market of meat products. The 2 diseases are enzootic in many parts of the world, such as Africa, southwest and central Asia, and the Indian subcontinent. Fever, generalized pocks, mucopurulent nasal discharge, and high mortality are the important clinical symptoms of the diseases. The causative agents, Sheeppox virus (SPPV) and Goatpox virus (GTPV), belong to the genus Capripoxvirus, subfamily Chordopoxvirinae, of family Poxviridae. Members of the Capripoxvirus genus are closely related, with genomic identities ranging from 96% (among species) to 99% (among isolates of the same species). 18,19 Capripoxviruses tend to be host specific; however, there are incidences of viruses crossing species barrier between goats and sheep. 3 Viruses can be detected in nasal secretions of naturally infected animals, as well as in lesions found in the upper and lower respiratory tracts of infected animals. 1
Comparative efficacy of different assays in the detection of Sheeppox virus and Goatpox virus from suspected clinical samples.*
Continued.

CIE = counterimmunoelectrophoresis; PCR = polymerase chain reaction; Ct = threshold cycle; CADRAD = Centre for Animal Disease Research and Diagnosis; IVRI = Indian Veterinary Research Institute; VBRI = Veterinary Biologicals and Research Institute; CSWRI = Central Sheep and Wool Research Institute; WRDDL = Western Regional Disease Diagnostic Laboratory; CIRG = Central Institute for Research on Goats; RDDL = Regional Disease Diagnostic Laboratory; ADRI = Animal Disease Research Institute; + = light band; ++ = moderately thick band; +++ = thick and bright band; – = sample was negative.
Control and eradication of capripoxvirus infections depend on accurate diagnosis and suitable prophylaxis. Therefore, a rapid, sensitive, and economic diagnostic tool for mass screening of affected sheep and goat flocks is essential. A routinely employed antigen detection conventional assay, like counterimmunoelectrophoresis (CIE), is less sensitive and less reproducible, and interpretation of results is more subjective than among molecular assays. Among molecular techniques, polymerase chain reaction (PCR) has been successfully used in the diagnosis of various infectious diseases and is considered one of the best alternatives to conventional assays because of its sensitivity, specificity, and reproducibility. 11,13 Conventional PCR assays have successfully been employed for routine screening of capripoxviruses, 7,12,13 but they are time consuming and require post-PCR manipulations, which sometimes lead to carryover contamination. 6,10 To address these drawbacks, many researchers use the rapid TaqMan probe-based quantitative PCR (qPCR) for the detection of orthopoxviruses, 8,16 GTPV, 2 SPPV, 1 and parapox-viruses. 5,15 The present study was aimed at developing both conventional and qPCR assays based on the highly conserved DNA polymerase (DNA pol) gene for the specific detection of capripoxviruses from clinical samples.
The following viruses were obtained from the poxvirus repository of the Division of Virology, Indian Veterinary Research Institute (IVRI; Mukteswar, India): SPPV-Srinagar isolate (passage 26), GTPV-Uttarakashi vaccine virus (passage 61), Orf virus (ORFV)-Mukteswar isolate (passage 47), Camelpox virus 1 (CMLV-1)-standard virus (passages 2–5), and Buffalopox virus (BPXV)-Vijayawada 96 vaccine virus (passage 50). These viruses were propagated in Vero (African green monkey kidney) cells a using Eagle minimum essential medium b supplemented with 10% newborn bovine calf serum. Infected Vero cells in 25-cm 2 tissue culture flasks showing greater than 80–90% cytopathic effect were harvested after decanting the media, and the cells were pelleted and resuspended in 250 μl of phosphate buffered saline (PBS; pH 7.2). The suspension was used for genomic DNA (gDNA) extraction.
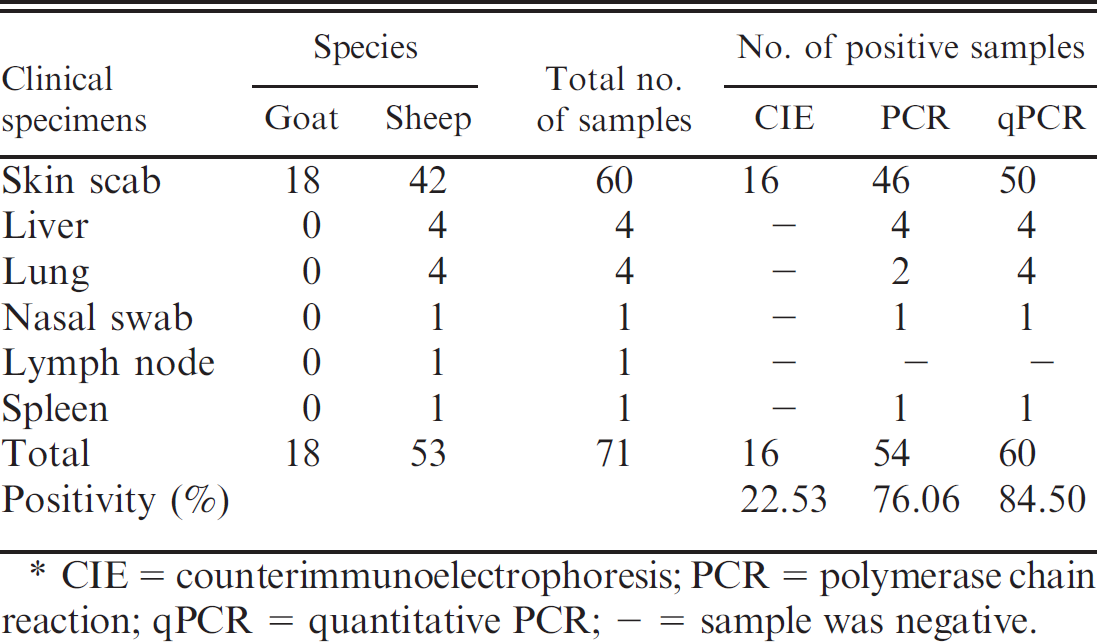
The details of the clinical samples used in the study for evaluation of the assays are summarized in Table 1. These samples were collected from goats and sheep suspected of capripox infections from different geographic areas of India and maintained in the virus repository. Scab and tissue materials were triturated in PBS, and a 10% (w/v) suspension was made for the extraction of gDNA.
Infected Vero cells suspended in 250 μl of PBS or 250-μl suspensions of suspected clinical samples were used for extraction of gDNA using a commercial kit. c The extracted DNA was dissolved in 50 μl of nuclease-free water (NFW) and stored at −20°C until further use. SPPV-Srinagar isolate (passage 26) and GTPV-Uttarakashi vaccine virus (passage 61) grown in Vero cells were purified on a 30–66% discontinuous sucrose density gradient and used to obtain concentrated viral DNA for standardization of the PCR assays. The purity and quantitation of DNA were assessed spectrophotometrically.
The oligonucleotide primers and probe were designed based on the published Capripoxvirus sequences in the GenBank database, made specifically to target the DNA pol gene, and were procured commercially. d The location of the primers/probes with respect to GTPV G20-LKV strain (GenBank accession no. AY077836) genome is indicated in the brackets of the nucleotide sequences. The PCR assays were standardized using positive and negative control DNA extracted from purified GTPV/SPPV and mock-infected Vero cells, respectively. Conventional PCR was performed in a 25-μl reaction mix containing 2 μl of DNA (approximately 420 ng), 10 pmol of each primer (forward HCPol 1: 5′-ACACCACCAACGCCTTTATGA-3′ [33870–33890] and reverse HCPol 2: 5′-TTACGGTATATTTCAAACAGATTAGAG-3′ [33977–34003]), 2.5 μl of 10× buffer, 10 μmol/l each deoxyribonucleotide triphosphate, and 0.25 IU of Taq DNA polymerase e in a thermal cycler. f The cycling conditions were initial denaturation at 95°C for 4 min and 35 cycles of denaturation at 94°C for 30 sec, annealing at 63°C for 30 sec, and extension at 72°C for 1 min, with a final extension of 7–10 min. An aliquot (5–10 μl) of PCR products was analyzed on 2% agarose gel electrophoresis to visualize the amplicons after staining with ethidium bromide.
Similarly, qPCR was carried out in a 25-μl reaction mixture containing 10 pmol of each primer (FP3: 5′-GGAATGATGCCRTCTARATTCCTATC-3′ [32922–32947] and RP3: 5′-CCCTGAAACATTAGTATCTGTATTTGTTGC-3′ [33072–33101]), and 5 pmol of TaqMan probe (fluorogenic hydrolysis [TaqMan] probe [P3] labeled with carbocyanin [Cy5] at the 5′ end and nonfluorescence Quencher TAMRA at the 3′ end: 5′-Cy5-CATCRCATCTAGGTTCRCAATGGATT-3′-TAMRA [32974–32999]), with other reagents similar to conventional PCR in a Mx 3000p qPCR machine. g The qPCR cycling conditions were initial denaturation at 94°C for 4 min and 40 cycles of denaturation at 94°C for 30 sec, and primer annealing and extension steps together at 58°C for 1 min. A standard curve was also performed for absolute quantification as well as to find out the sensitivity and efficiency ofthe assay using serial 10-fold dilutions of a purified viral DNA ranging from 10−2 (4.2 ng) to 10−8 (4.2 fg). The cycle threshold (Ct) values of the samples were recorded.
Analytic sensitivity of the PCR was determined using purified GTPV viral DNA (approximately 420 ng of viral DNA per 2 μl). Serial 10-fold dilutions of DNA ranging from 42 ng to 42 pg in 2 μl were prepared in NFW and tested using the 2 assays. The sensitivity was defined as the lowest quantity of viral DNA giving an amplification signal. The gDNA samples extracted from cell culture-adapted GTPV, SPPV, ORFV, CMLV, and BPXV isolates were tested to assess the diagnostic specificity. The specificity of the products was confirmed upon sequence analysis. Finally, the assays were applied on the DNA extracted from field clinical materials (n = 71) collected from sheep and goats suspected of pox infections.
Effective control of any disease necessitates a prophylactic as well as a specific and sensitive assay for diagnosis and epidemiologic studies. In the present study, conventional and TaqMan PCR assays based on the highly conserved DNA pol gene sequences to identify capripoxvirus from the suspected clinical samples were developed using different primer sets, which yielded a specific product of 134 bp and 180 bp in conventional PCR and qPCR, respectively.
In general, for routine diagnosis of a virus of different origin, PCR using specific primers targeting a highly conserved gene is most desirable to avoid false-negative results. Hence, in the current study the primers were designed based on the published Capripoxvirus sequences using the primer select program of Lasergene 6.0 h for the development of diagnostic PCR targeting the DNA pol gene, which is highly conserved among capripoxviruses and has significant sequence identity with other poxviruses (Sahay B: 2006, Evaluation of gene silencing by RNA interference (RNAi) in control of animal virus infections. PhD Thesis, Deemed University, IVRI, Bareilly, India). 14 The DNA pol gene is located in the central region of the genome and codes for the most conserved nonstructural protein. This protein plays a prime role in viral replication. 9 The phylogenetic analysis of the gene sequence of the Capripoxvirus 18 genus has shown that the DNA pol gene is group specific and highly conserved across the genus.
The sequence analysis of the DNA pol gene for primer selection revealed that designing specific Capripoxvirus primers is not straightforward because of adenine and thymidine (AT)-rich genomic regions in comparison with the genomes of other mammalian poxviruses of the Chordopoxvirinae subfamily. However, the conventional and TaqMan PCR assays resulted in the expected 134-bp and 180-bp products, respectively (data not shown), when applied on DNA from cell culture-derived GTPV and SPPV, indicating the specificity of the designed primers for the detection of capripoxvirus nucleic acid. In spite of 2 putative base mismatches in the TaqMan FP3 primer with reference to the SPPV genome, the qPCR assay was still able to detect SPPV, most likely because these mismatches are not located at the 3′ end of the primer.
The concentrations of each ingredient of the master mix and thermal profiles of both PCR assays were optimized to achieve maximum efficiency of amplification and specificity of reactions. The conventional PCR was carried out at annealing temperatures (Ta) ranging from 58°C to 66°C. The remaining thermoprofile steps were constantly maintained throughout the assay. Specific amplification was achieved at Ta up to 64°C; beyond this temperature, the intensity of amplification steadily decreased (data not shown). An optimum Ta of 63°C was adopted because it yielded an amplification efficiency of 99.5% (Y = −3.333 logX + 29.63; R 2 = 0.983) when tested using the SYBR Green dye commercial qPCR kit i (data not shown). The same conventional PCR reaction and cycling conditions were followed for SYBR Green dye-based PCR and the serial 10-fold dilutions of a viral DNA employed for determination of the standard curve.
When various Ta were used in the qPCR, a Ta of 58°C was considered optimum, with an amplification efficiency (Y = −3.361 logX + 23.26) of 98.4%. The efficiency (10−1/slope) of amplification (100.297) was approximately 1.98, with an R 2 value of 0.999 (Fig. 1A) in the standard curve, indicating the doubling of expected product after each cycle. The qPCR assay amplified only capripoxviruses (SPPV and GTPV; Fig. 1B) and was 100 times more sensitive than conventional PCR because it could detect as little as 0.042 pg of purified viral DNA (Fig. 1C). The samples that did not yield any Ct value within 35 cycles were considered negative for capripoxviruses because the fluorescence remained static or below the base level. The fluorescence remained static even for known positive samples (GTPV/SPPV) when they were used at higher dilutions, indicating the absence of virus-specific amplicons.
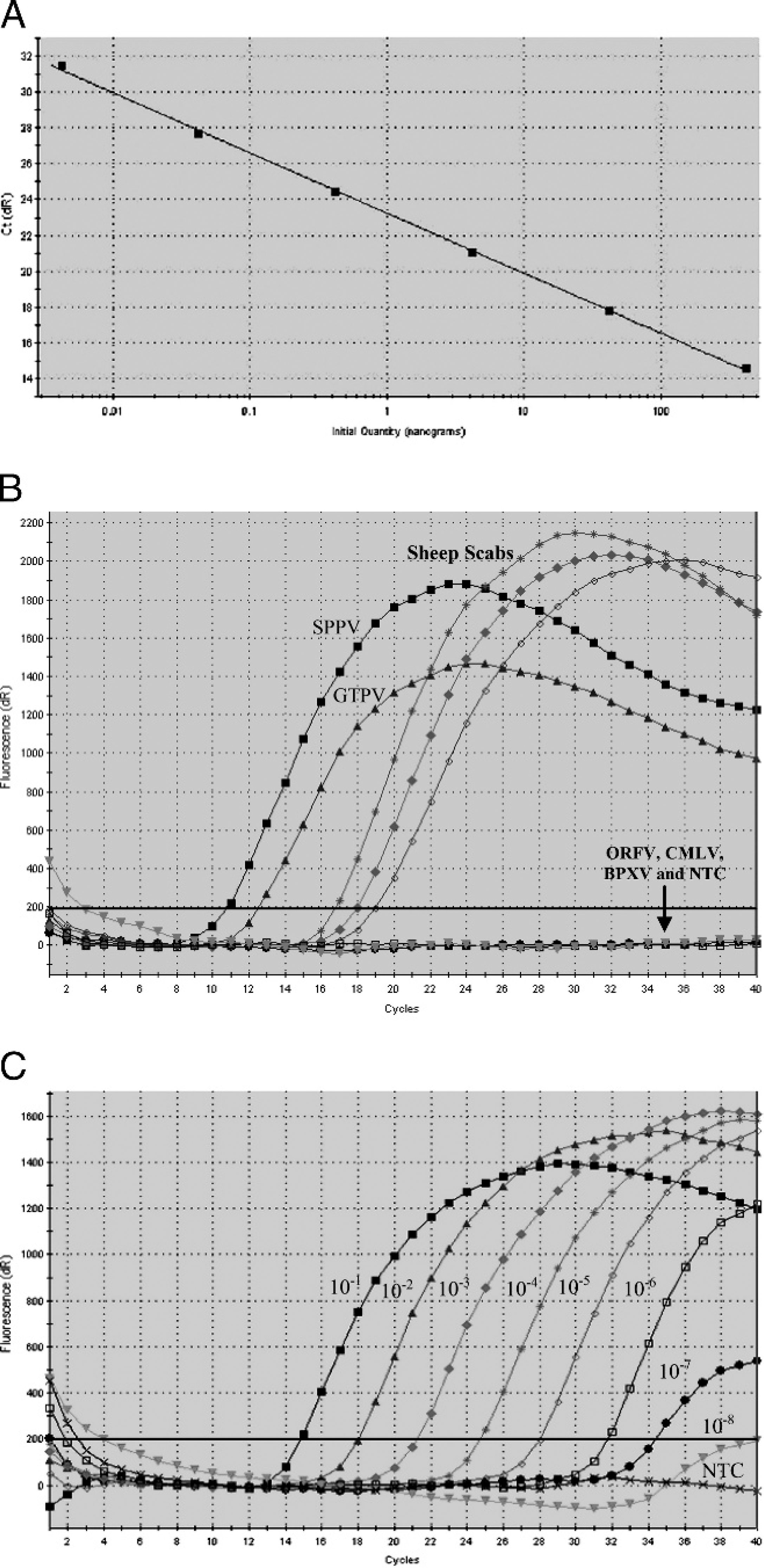
TaqMan probe (DNA pol gene)–based quantitative polymerase chain reaction.
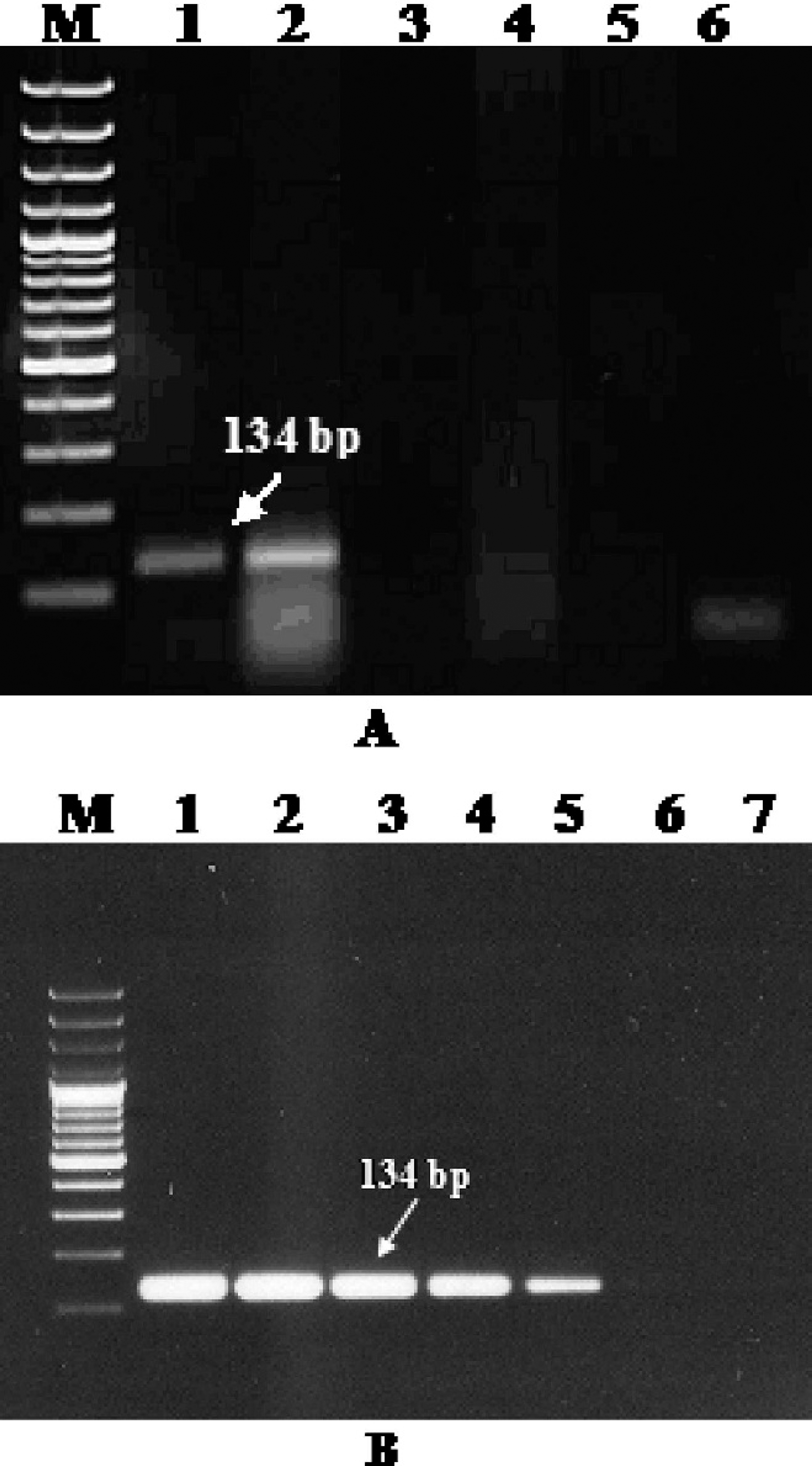
Specificity and sensitivity of conventional polymerase chain reaction.
When DNA was extracted from 5 poxviruses (GTPV, SPPV, ORFV, CMLV, and BPXV) tested, only capripox-viruses (SPPV and GTPV) were amplified. The specific size of amplicons was observed in a 2% agarose gel (Fig. 2A), and the specificity of partial DNA pol gene product was further confirmed upon cloning and sequencing. For this purpose, the amplicons were purified using a commercial gel extraction kit, j cloned into pGEM-T Easy vector, k and sequenced in an automated DNA sequencer. l The conventional and qPCR products (134 nucleotides and 180-nucleotide length of the GTPV and SPPV) of DNA pol were identified (GenBank accession nos. EU344751 and EU344752), and sequence analysis showed 100%, 100%, and 98%, and 96%,97%, and 100% nucleotide identity with published GTPV, Lumpy skin disease virus (LSDV), and SPPV isolates, respectively. The limit of detection of the conventional PCR assay was 4.2 pg (Fig. 2B).
Results of suspected clinical samples tested for the detection of capripoxvirus by different assays.*
CIE = counterimmunoelectrophoresis; PCR = polymerase chain reaction; qPCR = quantitative PCR; –= sample was negative.
Recently, a highly sensitive and specific Poly A polymerase gene-based TaqMan PCR assay based on a commercial kit has been reported for rapid detection of SPPV in the preclinical (nasal swab and buffy coat), clinical (scab materials), and postmortem (lymph nodes and lungs) samples collected from lambs experimentally infected with a virulent strain of SPPV. 1 However, the authors also suggested testing clinical materials suspected of LSDV from cattle and GTPV from goats to further confirm the diagnostic effectiveness of that assay. 1 The qPCR assay based on the DNA pol gene was developed to detect capripoxvirus from the suspected clinical specimens, which do not use a commercial kit. The standardized qPCR in the present study using purified viral DNA of SPPV and GTPV was easily adopted for direct detection of capripoxvirus DNA in clinical field samples.
Routinely, CIE has been carried out for diagnosis of sheep and goat infections in the authors' laboratory. 17 The results of testing 71 clinical samples by CIE, conventional PCR, and qPCR (Table 2) reveal that 54 (76.06%) and 60 (84.50%) were positive in conventional and TaqMan assays, respectively. These results indicate that both assays are adequate for the detection of sheeppox and goatpox viruses from skin scab, lung, liver, nasal swab, and spleen samples. The results also showed higher diagnostic efficacy of TaqMan PCR than conventional PCR. The extent of agreement between the results of both assays was high, indicating the virus load in the clinical samples is sufficient enough for the detection of the target gene by both assays.
Lumpy skin disease virus was not included in the present study because to the authors' knowledge, this disease does not occur in India. Nevertheless, it is presumed that the assays would work for LSDV DNA because the conventional PCR and qPCR primers sequences are, respectively, 100% and 92% identical with published LSDV sequences. The less-than-perfect identity of the qPCR primers is due to a 2-nucleotide mismatch in the middle of the primer sequences, which is similar to the mismatch with SPPV sequences. Because the 2-nucleotide mismatch did not adversely affect the performance of the qPCR on SPPV, a similar situation should prevail with the LSDV.
The qPCR assay reported in the present study was found to be rapid (it requires only 1 hr 40 min vs. 2 hr in conventional PCR, with additional time required for manipulation of post-PCR products in agarose gel), specific, highly sensitive, and economic (in terms of cost, because no commercial qPCR kit is used), and is a suitable screening tool for confirmatory detection of capripox-viruses from clinical material of goatpox and sheeppox suspect cases. Moreover, in conventional PCR, qualitative analysis is an endpoint analysis after the PCR has reached the plateau phase and is not as accurate as quantitative. In qPCR, the PCR can be analyzed at the log-linear phase of each cycle. The qualitative analysis of the conventional PCR is gel based and provides a visual assessment of specificity and quality of the PCR amplification.
The qPCR assay represents an improvement over earlier methods of capripox detection because of its rapidity, simplicity, and ability to be carried out in laboratories without the need for cell culture facilities. The qPCR assay is now regularly used along with virus isolation for routine screening of sheeppox- and goatpox-suspected clinical samples received at the authors' laboratory for confirming outbreaks. The assay has the potential to be used as a replacement or complementary test with conventional assays for diagnosis of capripoxviruses. To explore the potential of this assay, it is desirable to evaluate it with LSDV to establish its specificity and sensitivity for all members of the Capripoxvirus genus.
Acknowledgements. The authors thank the Director of the Indian Veterinary Research Institute for providing the necessary facilities to carry out this work. This study was supported by grants from the Ministry of Forest and Environment of the Indian government under the All India Coordinated Project on the Taxonomy capacity building of poxviruses. Vinayagamurthy Balamurugan and Kallesh Danappa Jayappa contributed equally to this publication.
Footnotes
a.
ATCC CCL81, American Type Culture Collection, Manassas, VA.
b.
Sigma-Aldrich, St. Louis, MO.
c.
AuPreP™ DNA Extraction Kit, Life Technologies Pvt. Ltd., New Delhi, India.
d.
Integrated DNA Technologies Inc., Coralville, IA.
e.
Invitrogen Corp., Carlsbad, CA.
f.
PTC 200 Thermal Cycler, MJ Research Inc., Waltham, MA.
g.
Mx3000p® QPCR System, Stratagene Inc., La Jolla, CA.
h.
DNASTAR Inc., Madison, WI.
i.
Qiagen Inc., Valencia, CA.
j.
MinElute® Gel Extraction Kit, Qiagen Inc., Valencia, CA.
k.
pGEM®-T Easy Vector System, Promega Corp., WI.
l.
ABI PRISM® 3100, Applied Biosystems, Foster City, CA.