
Abstract
Feline immunodeficiency virus (FIV) is an important infectious agent of cats. Clinical syndromes resulting from FIV infection include immunodeficiency, opportunistic infections, and neoplasia. In our study, a 5′ long terminal repeat/gag region–based reverse transcription insulated isothermal polymerase chain reaction (RT-iiPCR) was developed to amplify all known FIV strains to facilitate point-of-need FIV diagnosis. The RT-iiPCR method was applied in a point-of-need PCR detection platform—a field-deployable device capable of generating automatically interpreted RT-iiPCR results from nucleic acids within 1 hr. Limit of detection 95% of FIV RT-iiPCR was calculated to be 95 copies standard in vitro transcription RNA per reaction. Endpoint dilution studies with serial dilutions of an ATCC FIV type strain showed that the sensitivity of lyophilized FIV RT-iiPCR reagent was comparable to that of a reference nested PCR. The established reaction did not amplify any nontargeted feline pathogens, including Felid herpesvirus 1, feline coronavirus, Feline calicivirus, Feline leukemia virus, Mycoplasma haemofelis, and Chlamydophila felis. Based on analysis of 76 clinical samples (including blood and bone marrow) with the FIV RT-iiPCR, test sensitivity was 97.78% (44/45), specificity was 100.00% (31/31), and agreement was 98.65% (75/76), determined against a reference nested-PCR assay. A kappa value of 0.97 indicated excellent correlation between these 2 methods. The lyophilized FIV RT-iiPCR reagent, deployed on a user-friendly portable device, has potential utility for rapid and easy point-of-need detection of FIV in cats.
First isolated from a domestic cat in California in 1986, 13 Feline immunodeficiency virus (FIV) is a pathogen of domestic cats worldwide. FIV, a close relative of Human immunodeficiency virus 1 and 2 (HIV) in both immunological and pathogenic properties, is also a member of the genus Lentivirus of the family Retroviridae. 3 Based on genetic variations found in its env V3-V4 region and gag gene and geographic location, FIV has been grouped into multiple clades. 8 Signs include an acute flu-like illness and lymphadenopathy, followed by a long subclinical period during which the virus replicates at low levels. Cluster of differentiation (CD)4+ lymphocyte numbers and the CD4-to-CD8 ratio decline gradually, accompanied by progressive weakening of immune function. 6 During acute and chronic stages of infection, although antibody levels are variable from cat to cat, significant levels of viremia and/or proviral DNA are present in white blood cells or other sites. 22
Laboratory tests are required for FIV disease diagnosis because clinical signs caused by FIV infection and by other immunodeficiency diseases, such as Feline leukemia virus infection, are quite similar. In addition, FIV-infected cats appear healthy for many years. Although a reliable diagnostic tool, isolation of FIV from peripheral blood lymphocytes 11 is time- and labor-intensive, and impractical for routine use. Antibodies against FIV are detectable within 2–6 weeks after infection. Many animal shelters use FIV antibody enzyme-linked immunosorbent assays (ELISAs) to screen cats for FIV before making a decision on eligibility for adoption or euthanasia. Diagnosis of FIV can literally be a life or death result for millions of cats; thus, diagnostic accuracy is of the highest importance. 5 However, antibody response is generally misleadingly low during the first 2–4 weeks post-infection, and in some cases up to a year, 15 as well as during the terminal stages of infection. Moreover, vaccination with a commercially available killed whole-virus vaccine 5 and the presence of maternal anti-FIV antibodies in very young animals can lead to false-positive results, making interpretation of serological results problematic.
Nucleic acid amplification–based methods, usually more sensitive and specific in detecting the target pathogen than immunoassays, are an alternative to detect FIV infection. PCR-based assays targeting conserved regions in pol, gag, and/or 5′ untranslated region (5′-UTR)/gag regions of the FIV genome have been developed to aid FIV diagnosis. 7 Low numbers of virus-infected cells in the blood during the lengthy subclinical phase of infection, when most cats are tested, make detection difficult. Highly sensitive PCR assays, such as real-time PCR (qPCR), provide improved detection for use as an FIV confirmatory test in cats with a positive antibody test result. 1 In addition, molecular amplification of FIV genome and/or proviral DNA could be used to distinguish natural infection from vaccination. 12 However, these tests tend to be limited to commercial and research diagnostic laboratories because of the requirement of sophisticated equipment and trained technicians. 2 Point-of-need or point-of-care (PON/POC) FIV testing helps shorten test turnaround time and eliminate shipping and processing costs. A rapid, sensitive, user-friendly FIV nucleic acid detection test that could facilitate easy PON/POC agent detection and disease management would be of benefit to the veterinary community.
A user-friendly and portable PCR system a for animal PON/POC testing has been developed. This system relies on fluorescent probe hydrolysis–insulated isothermal PCR (iiPCR),4,16,17 which is based on convection PCR. 10 The 3 stages (denaturation, annealing, and extension) of PCR are achieved by cycling the reaction components through different temperature zones during convection. The reaction and data processing steps are completed without post-amplification processing in less than 1 hr. Several assays based on this system have been demonstrated to provide sensitivity and specificity equivalent to those of qPCR or nested PCR (nPCR).2,18,19,21
In our study, we developed and evaluated an RT-iiPCR method for detection of FIV for PON/POC applications. Performance of the established assay to detect FIV in feline clinical samples was compared with a FIV nPCR assay 1 targeting the pol gene of the FIV provirus using degenerate primer sets.
The FIV RT-iiPCR was designed to amplify a conserved area of the 5′ long terminal repeat (5’-LTR)/gag region of FIV found in 76 entries in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). Primers and probes (Table 1) were designed b by following principles suggested for iiPCR (http://www.iipcr.com/eweb/uploadfile/20130522114104277.pdf). Analysis was performed with Mfold (http://mfold.rna.albany.edu/?q=mfold) to avoid amplicons with major secondary structures. Combinations of primers and probes, and concentrations of primers, probe, deoxyribonucleotide triphosphates, Moloney Murine leukemia virus reverse transcriptase, and Taq DNA polymerase were optimized and the reaction mixture freeze-dried 14 and stored at room temperature. After reconstituting the reaction components with 50 μL of premix buffer, c 5 μL of sample nucleic acids were added to the reaction. Subsequently, 50 μL of the final mixture were transferred to an R-tube, c which was next sealed with a cap, spun for 10 sec, d and placed into the portable PCR machine a to run the reaction.
Primers and probes used in the current study.

Nucleotide position is based on GenBank accession M25381.
RT-iiPCR = reverse transcription insulated isothermal polymerase chain reaction.
The program of the portable PCR machine a included a 42°C/10 min step for RT and a 95°C/30 min step for iiPCR. The reaction was completed in 1 hour. The results were determined as “+,” “–,” or “?” according to the default signal-to-noise (S/N) thresholds.4,17
To determine analytical sensitivity of the established assay, a synthetic plasmid containing the T7 promoter-LTR/gag region of the FIV genome cassette e was used to prepare RNA transcribed in vitro (IVT) RNA using a commercial kit. f After removal of residual DNA, g the concentration of RNA was determined with a spectrophotometer. h The concentration of the IVT RNA molecules per microliter was calculated according to the following formula:
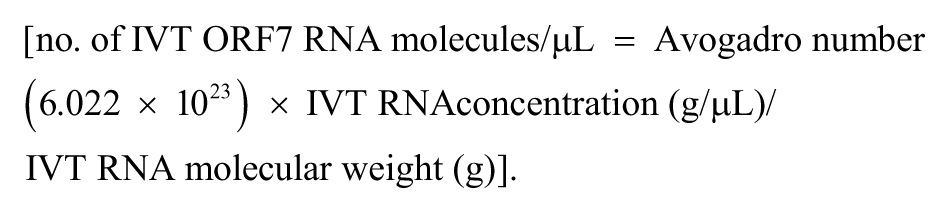
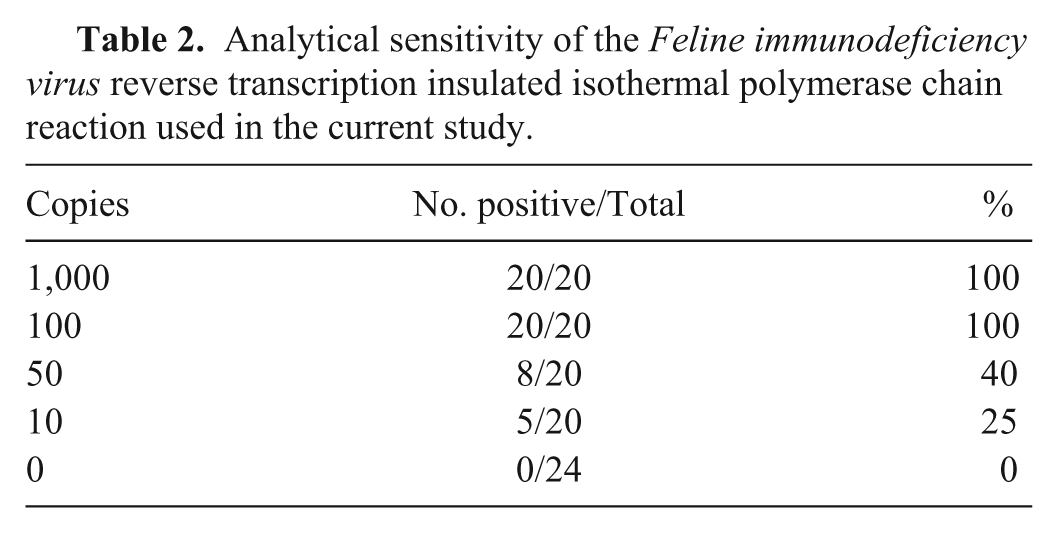
Aliquots of both the plasmid DNA and IVT RNA were prepared for single use and stored at -80°C. The detection threshold of the RT-iiPCR reaction was determined by amplification of serial dilutions (1,000, 100, 50, or 10 copies) of IVT RNA transcript made in 40 ng/μL of yeast transfer RNA. Based on the results (Table 2), limit of detection (LOD) 95% of FIV RT-iiPCR was determined to be ~95 copies RNA per reaction by statistical Probit analysis (a nonlinear regression model) using commercial software. i Intra- and interassay repeatability were evaluated by testing 6 replicates containing IVT RNA of ~5-fold LOD 95% (500 copies) in 3 runs using 3 different portable devices a (Table 3). Each run included a positive (plasmid DNA) and nontemplate control (double-distilled H2O). Positive results were obtained from all test samples (Table 3), indicating excellent reproducibility of the FIV RT-iiPCR assay.
Analytical sensitivity of the Feline immunodeficiency virus reverse transcription insulated isothermal polymerase chain reaction used in the current study.
Repeatability, intra-assay, and interassay performance evaluation of the Feline immunodeficiency virus reverse transcription insulated isothermal polymerase chain reaction used in the current study.*
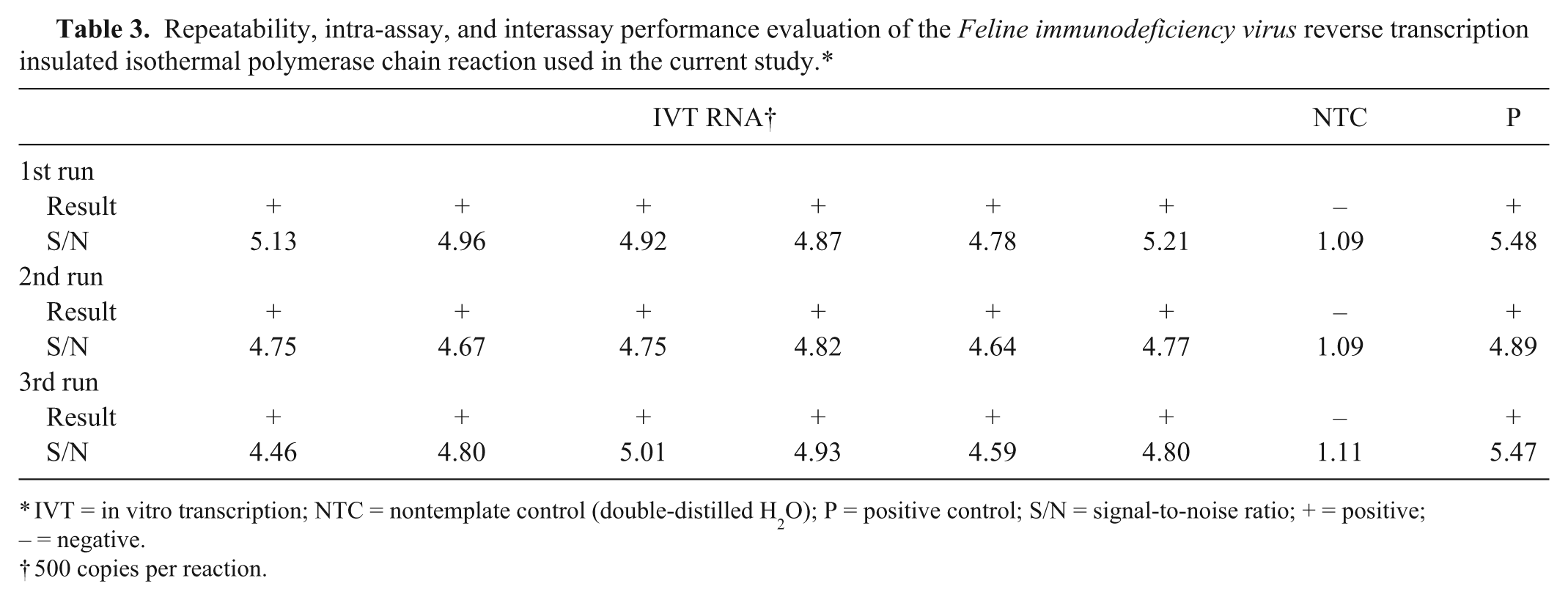
IVT = in vitro transcription; NTC = nontemplate control (double-distilled H2O); P = positive control; S/N = signal-to-noise ratio; + = positive; – = negative.
500 copies per reaction.
The performance of FIV RT-iiPCR in detecting FIV was evaluated by comparing limits of RT-iiPCR with a nPCR assay 1 in detecting a member of subtype B (FIV type strain j ), the predominant clade found in the United States. 20 Both steps of nPCR were performed with 50 µL total volume containing Taq polymerase mix, k 400 nM of each primer (Table 1), and 5 µL of DNA template in a thermocycler. l The cycling program for both runs was 3 min at 94°C, 45 cycles of 15 sec at 94°C, 30 sec at 50°C, and 45 sec at 72°C, and finally 10 min at 72°C. The assay resulted in a 576-bp product, which was detected by agarose gel electrophoresis. The side-by-side comparison was performed using 10-fold serial dilutions of the extracted nucleic acid m prepared from the FIV type strain. The results showed that detection endpoints were ~10−3 dilution of FIV nucleic acid for both RT-iiPCR and nPCR assays (Table 4), indicating that the 2 methods had similar analytical sensitivity in detecting FIV.
Comparison of detection limits between reverse transcription insulated isothermal polymerase chain reaction (RT-iiPCR) and nested PCR using serial dilutions of a Feline immunodeficiency virus type strain.*
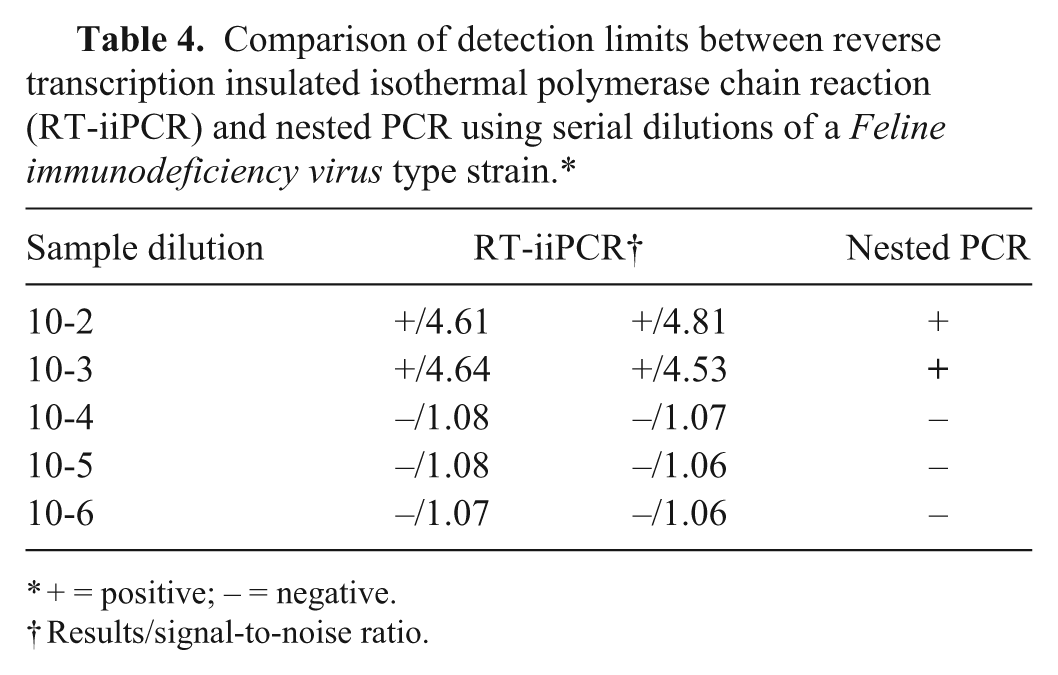
+ = positive; – = negative.
Results/signal-to-noise ratio.
An exclusivity panel including Felid herpesvirus 1 (FVR-SGE strain n ), feline coronavirus, o Feline calicivirus, p Feline leukemia virus, q Mycoplasma haemofelis, and Chlamydophila felis (nucleic acid from the University of Tennessee [UT] Diagnostic Laboratory, Knoxville, Tennessee) were used to evaluate analytical specificity of FIV RT-iiPCR. The assay gave negative results with all members in the exclusivity panel, indicating that this reaction could detect FIV with high specificity.
Finally, the performance of FIV RT-iiPCR in detecting FIV in clinical samples was evaluated. For this purpose, 76 clinical samples (whole blood and bone marrow in ethylenediamine tetra-acetic acid), submitted to the University of Tennessee, College of Veterinary Medicine, Clinical Immunology Laboratory (Knoxville, Tennessee) from 2008 to 2014 for FIV testing, were evaluated with the RT-iiPCR assay. Nucleic acid had been extracted from clinical samples according to the manufacturer’s instructions. m The nucleic acid samples were stored at -80°C. The samples had been tested by the UT Diagnostic Laboratory with the nPCR method described previously. Any questionable results were confirmed by direct PCR product sequencing r following product clean-up. s Some of the samples were identified as subtypes B and C by sequencing (data not shown). Test results show that among the 45 samples that tested positive in the nPCR assay, RT-iiPCR generated detectable signals from 44 samples. All 31 nPCR-negative samples also tested negative by RT-iiPCR. Performance comparison between FIV nPCR and FIV RT-iiPCR was determined. The degree of agreement between the 2 assays was assessed by calculating kappa values with 95% confidence intervals. Compared with the nPCR, test sensitivity was 97.78% (44/45), specificity was 100.00% (31/31), and agreement was 98.65% (75/76), for the FIV RT-iiPCR. A kappa value of 0.97 indicated excellent correlation between these 2 methods.
The sample with the discrepant result (nPCR-positive and RT-iiPCR–negative) was submitted to an outside laboratory t for testing by qRT-PCR. The sample tested positive by the outside lab and was typed as a B2/E strain. This sample was also subjected to sequencing analysis of a PCR fragment of the env V3-V4 region using primers (Table 1) described previously. 9 BLAST (http://blast.ncbi.nlm.nih.gov) analysis of its nucleotide sequence showed that its sequence had the highest identity (97%) with a subtype B strain (GenBank accession U57019). Furthermore, the RT-iiPCR primers and probe binding regions of this sample were amplified by nPCR protocol with primers amplifying LTR and gag regions (Table 1). The cycler conditions for the first reaction were 94°C for 2 min, followed by 35 cycles of 94°C for 20 sec, 57°C for 30 sec, 72°C for 1.5 min, and a final extension step of 72°C for 7 min. The conditions for the second reaction were 94°C for 2 min, followed by 35 cycles of 94°C for 20 sec, 50°C for 20 sec, and 72°C for 30 sec. The nested product was purified s and directly sequenced r in both directions using the nested primer set. One and 3 mismatches were found in the forward and reverse primers, respectively. The reagent designed for RT-iiPCR was further tested for its ability to detect FIV in the same set of clinical samples using a real-time PCR machine. u The lyophilized primers and probe were suspended in 50 µL of the premix buffer. The samples were run in 12-µL total volume reactions using 10 µL of the reaction mix and 2 µL of nucleic acid. Positive and negative controls used for testing were as previously described. The cycler parameters were 92°C for 30 min, followed by 45 cycles of 93°C for 15 sec and 60°C for 60 sec. Performance comparison between use of the reaction mix in the portable PCR machine a versus the real-time PCR machine u resulted in 100% sensitivity (44/44) and 100% specificity (32/32) for the 2 machines.
Overall, the FIV RT-iiPCR performed with analytical sensitivity and specificity equivalent to that of the published nPCR method. The iiPCR assay was run with a RT step in the portable PCR machine a to provide optimal clinical sensitivity by amplifying both genomic RNA and proviral DNA forms of FIV. However, the assay can also be run without the RT for detection of proviral DNA only (as was done with the reagent in the real-time PCR machine), with a sensitivity of 5 copies of DNA per reaction (data not shown). This is important for recently vaccinated cats or cats with an unknown vaccination history. Presumably, FIV RNA derived from the vaccine will be quickly removed from circulation without integration of proviral DNA into the cat’s genome. Thus, proviral DNA should be detectable only in infected cats, but not in vaccinated cats. This hypothesis was confirmed in a previous study. 22 However, several commercially available molecular assays for FIV are RT-PCR assays, which detect both viral RNA and proviral DNA, potentially resulting in false-positive results for vaccinated cats.
Limited by the fact that no medical history was available for the clinical samples evaluated in our study, test results could not be evaluated based on defined populations of cats (i.e., vaccinated vs. naturally infected); nor was virus isolation or ELISA used for comparison. Therefore, although the sensitivity and specificity of the iiPCR-based assay are essentially the same as the nPCR protocol, the true test sensitivity and specificity (ability to detect infected vs. uninfected vs. vaccinated cats) are unknown and a limitation of the study. Additionally, comparison of results from this test with a gold standard method was not possible. Isolation of FIV is difficult, and there is no agreement on what test should be considered the gold standard for FIV infection status determination. 2
High sequence variation makes reliable PCR detection of FIV infection difficult. 5 The nPCR is based on degenerate primers targeting the pol gene of the FIV genome to cover known FIV strains with marked sequence variation. Although the RT-iiPCR consisted of nondegenerate primers and probe, its sensitivity and specificity in detecting FIV in clinical samples were comparable to those of the nPCR in detecting FIV in clinical samples. The primer and probe set of the iiPCR method targets a region with minimal sequence diversity (usually a single mismatch within a target area, but up to 3 mismatches in a single target area) among known FIV sequences. Most of the mismatches are located close to the 5′-end of the sequences, and these types of mismatches have been shown to be substantially tolerated with the iiPCR assay with sequenced strains from Taiwan (data not shown). Furthermore, data obtained during the development of iiPCR showed that the reaction is able to tolerate more mismatches than conventional PCR. This is likely because the components continuously cycle through temperature gradients in a cylinder vessel; the primer annealing step in iiPCR is not restricted to a specific temperature within the temperature gradients. This allows the primers to hybridize with sequences with a certain degree of mismatches, enhancing the sensitivity of the reaction. Apparently, clinical specificity of FIV RT-iiPCR was not compromised, as the assay did not generate any signals from Feline leukemia virus, also a member of the Retroviridae family.
Notably, the one sample generating discrepant results between the 2 tests was grouped with subtype B based on sequencing analysis of the env V3-V4 region. Primer Blast analysis of the FIV gag gene sequences available in the GenBank database revealed that this is the first known subtype B sequence with 3 mismatches to the reverse primer of iiPCR. This sample also contained very few virus copies (estimated 100 copies by qRT-PCR). Therefore, it is likely that the mismatches affected the assay sensitivity, limiting the detection limit for this sample. However, it is unknown if the iiPCR assay would have detected this virus if it had been present in a larger quantity in the sample. Sequence divergence of FIV will be monitored continuously in the future to evaluate the clinical significance of this strain.
A critical limitation of PCR for clinical diagnosis is cross-contamination caused by previously amplified products, which is particularly true of nPCR assays. Detection of fluorescent signals derived from hydrolysis of the probe of the portable system a helps reduce cross-contamination risks. In addition, automatic interpretation of the S/N ratios allows a technician with basic training to complete the test.
The reagent developed for use in the portable system a provided the same results when tested with clinical samples in a real-time PCR machine, making the use of these reagents possible in facilities with qRT-PCR capabilities without the use of the portable system. a However, methods (such as iiPCR with the portable system a ) that offer portability and automatic data interpretation can be applied to PON/POC FIV testing to facilitate timely disease management, pathogen identification in veterinary clinics, and customs inspection at ports of entry.
Footnotes
Acknowledgements
We thank Mrs. Rupal Brambhatt for technical support.
Authors’ contributions
RP Wilkes, YL Tsai, PYA Lee, HH Chang, LJ Ma, and HTT Wang contributed to conception and design of the study. HFG Chang contributed to conception of the study. RP Wilkes, YL Tsai, PYA Lee, and HH Chang contributed to acquisition, analysis, and interpretation of data. SA Kania contributed to analysis and interpretation of data. LJ Ma and HTT Wang contributed to interpretation of data. RP Wilkes, PYA Lee, and LJ Ma drafted the manuscript. RP Wilkes, SA Kania, PYA Lee, HFG Chang, and HTT Wang critically revised the manuscript. RP Wilkes, PYA Lee, and HTT Wang gave final approval. RP Wilkes and HTT Wang agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
POCKIT Nucleic Acid Analyzer, GeneReach USA, Lexington, MA.
b.
Primer Express software, Applied Biosystems, Foster City, CA.
c.
GeneReach USA, Lexington, MA.
d.
Cubee, GeneReach USA, Lexington, MA.
e.
Generay Biotechnology, Shanghai, China.
f.
Ambion MAXIscript T7 kit, Applied Biosystems, Foster City, CA.
g.
Ambion Turbo DNA-free kit, Applied Biosystems, Foster City, CA.
h.
NanoDrop 1000, NanoDrop Technologies, Houston, TX.
i.
SPSS Inc., Chicago, IL.
j.
NCSU1 (VR-2333) American Type Culture Collection, Manassas, VA.
k.
Premix Taq, Clontech Laboratories Inc., Mountain View, CA.
l.
GenePro PCR, BIOER Technology, Hangzhou, China.
m.
DNeasy blood and tissue kit, Qiagen Inc., Valencia, CA.
n.
FVR-SGE, U.S. Department of Agriculture, National Veterinary Services Laboratory, Ames, IA.
o.
WSU 79-1683 (VR-989), American Type Culture Collection, Manassas, VA.
p.
F-9 (VR-782), American Type Culture Collection, Manassas, VA.
q.
ST-FeLV (VR-719), American Type Culture Collection, Manassas, VA.
r.
Molecular Sequencing Facility, University of Tennessee, Knoxville, TN.
s.
Exo-SAP-IT, Affymetrix Inc., Santa Clara, CA.
t.
Molecular Diagnostics Lab, Pathobiology Diagnostic Services, Auburn University, Auburn, AL.
u.
Applied Biosystems Step One real-time PCR system, Life Technologies, Grand Island, NY.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Y-L Tsai, P-YA Lee, H-H Chang, L-J Ma, H-FG Chang, and H-W Thomas are affiliated with GeneReach USA.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.