
Abstract
Sporadic outbreaks of vesicular stomatitis (VS) in the United States result in significant economic losses for the U.S. livestock industries because VS is a reportable disease that clinically mimics foot-and-mouth disease. Rapid and accurate differentiation of these 2 diseases is critical because their consequences and control strategies differ radically. The objective of the current study was to field validate a 1-tube multiplexed real-time reverse transcription polymerase chain reaction (real-time RT-PCR) assay for the rapid detection of Vesicular stomatitis New Jersey virus and Vesicular stomatitis Indiana virus strains occurring in Mexico and North and Central America. A comprehensive collection of 622 vesicular lesion samples obtained from cattle, horses, and swine from throughout Mexico and Central America was tested by the real-time RT-PCR assay and virus isolation. Overall, clinical sensitivity and specificity of the real-time RT-PCR were 83% and 99%, respectively. Interestingly, VS virus isolates originating from a specific region of Costa Rica were not detected by real-time RT-PCR. Sequence comparisons of these viruses with the real-time RT-PCR probe and primers showed mismatches in the probe and forward and reverse primer regions. Additional lineage-specific primers and a probe corrected the lack of detection of the missing genetic lineage. Thus, this assay reliably identified existing Mexican and Central American VS viruses and proved readily adaptable as new VS viruses were encountered. An important secondary result of this research was the collection of hundreds of new VS virus isolates that provide a foundation from which many additional studies can arise.
Introduction
Vesicular stomatitis (VS), a disease of cattle, horses, and swine, is characterized by severe vesiculation and/or ulceration of the tongue, oral tissues, feet, and teats and substantial loss of productivity. The disease is notable because VS in cattle and swine is clinically indistinguishable from foot-and-mouth disease (FMD), a problem that requires different control strategies and entails enormous economic loss. Outbreaks of VS also result in significant economic loss by reducing productivity and triggering animal and animal-product quarantines, but VS outbreaks are self-limiting. Unlike FMD, VS spreads readily to humans, resulting in a transient but often incapacitating illness. Vesicular stomatitis is endemic in southern Mexico, Central America, and northern South America. Epidemic VS has occurred 22 times in the United States over the past 120 years. Recent epizootics in the United States have involved hundreds of farms and often encompass several southwestern states. 6,15
The goal of the present study was to test a polymerase chain reaction (PCR) assay designed to identify field isolates of VS virus; the details of design and bench validation of this test are the subject of a separate article. This seemingly simple goal was complicated by 3 factors. First, VS epizootics in the United States are caused by 2 distinct serotypes of VS virus, Vesicular stomatitis New Jersey virus (VSNJV) and Vesicular stomatitis Indiana virus (VSINV), both members of the order Mononegavirales, family Rhabdoviridae, and genus Vesiculovirus. The 2 serotypes differ by about 30–70% at the nucleic acid level, and these differences are spread across the genome. 13 Second, these viruses appear to originate from southern Mexico and spread during the summer to the southwestern United States, where they initiate each epizootic de novo. 15,17 Each epizootic is initiated by a distinct virus, which then evolves as the epizootic expands during the summer and autumn. 15,17 Central American VS virus isolates exhibit an unusual genetic heterogeneity that varies with latitude and altitude rather than time. 10,18 In addition, the full extent of animal-to-animal and year-to-year sequence variation at any specific location is unknown. It is not clear why one virus rather than another moves north to the United States to initiate an epizootic. Third, VS virus replicates as a quasi-species, a dynamic equilibrium of related sequence variants that adapt quickly to external factors. 11,21 To the authors' knowledge, a single PCR assay has never been developed for such a diverse population of viruses.
Materials and methods
Sample collection, shipment, and storage
The present research rested on the success of the Central American Screwworm Eradication Program. Screwworm eradication workers were assigned to visit farms in Central America every week to sample not only screwworm-suspect cases but also cases of vesicular disease. The samples were forwarded to the Vesicular Disease Diagnostic Laboratory (LADIVES, Panama City, Panama), where they tested negative for FMD before being released for VS virus studies. Thus, the samples available for the current research represented vesicular lesions occurring on livestock in 7 countries over a 2-year period. This represents a uniquely saturated sampling of a natural genetically diverse viral population. Most of the samples used in the present study came from Costa Rica and Mexico, where the assay was tested in both field and laboratory conditions. In addition, the assay was performed at the LADIVES laboratory on samples submitted from Central American countries as part of routine surveillance. Testing samples from such a broad geographic and genetic origin by real-time reverse transcription PCR (RT-PCR) showed that this developed test could identify all known VS virus variants causing disease and established the principle that a carefully designed PCR can reliably identify all characterized members of a genetically diverse natural viral population.
Central America. Vesicular lesion samples obtained from cattle, horses, or swine were placed in sterile glass vials containing approximately 20 ml of 50% phosphate buffered glycerin and transported in coolers to the appropriate laboratory. Samples throughout Central America were sent to LADIVES. Samples from Costa Rica were collected in duplicate; one of the duplicate samples was sent to LADIVES for diagnosis, and the second duplicate was transferred to 2.5-ml cryovials and kept at −80°C at the Universidad Nacional School of Veterinary Medicine (Heredia, Costa Rica) until processing.
Mexico. Clinical samples were obtained from Mexico's Department of Agriculture Exotic Animal Disease Commission (EADC), the responsible agency for investigating foreign animal diseases in Mexico. The EADC veterinarians from the States of Chiapas, Veracruz, and Tabasco carried out investigations when suspected cases of vesicular disease were reported and collected epithelium samples when vesicular lesions were observed. Epithelium samples were collected in duplicate and placed in sterile brown glass vials containing approximately 20 ml of 50% phosphate buffered glycerin and transported in coolers containing ice packs to the central regional EADC office in Tuxtla Gutierrez. Samples were in transit 1 12 hr depending on the distance of the affected premise to the EADC office. At the regional office, part of each epithelium sample was removed, placed in 2-ml cryovials containing 0.5 ml of sterile minimal essential medium (MEM) antibiotic and stored at −70°C until tested. Original samples were repacked with fresh ice packs and sent by air courier to the EADC Central Laboratory in Mexico City where routine diagnostic tests were carried out. The following information was recorded for each outbreak: year and month of the investigation, state, municipality, herd size, number of cases, number of fatalities, laboratory diagnosis, and species of animals.
Sample processing for PCR
Frozen epithelial samples were thawed in a water bath at 37°C and then kept on ice for less than 1 hr. Tissue was removed from the vial and washed 3 times with phosphate buffered saline (PBS). The RNA was isolated using a commercially available guanidine thiocyanate–based extraction kit a as per the manufacturer's instructions. Briefly, approximately 30 mg (or half of smaller samples) was added to 560 μl of lysis buffer in a 1.5-ml microcentrifuge tube, disrupted with a sterile pestle, b and centrifuged for 1 min at 10,000 × g. Six hundred microliters of the supernatant was transferred to a disposable cell-lysate homogenizer c and centrifuged at 10,000 × g for 2 min. To the flow-through was added 700 ul of 70% ethyl alcohol, and the mixture was centrifuged through a silica-based spin column. a The column was washed, and RNA was eluted in 40 μl of water as prescribed in the manufacturer's instructions.
Sample processing for virus isolation
Approximately 30 mg or half of smaller samples was added to 1 ml of medium consisting of Dulbecco's modified Eagle's medium d with 4% fetal bovine serum e and antibiotics (200 U penicillin, d 200 μg streptomycin, d and 0.5 μg amphotericin d B/ml) and macerated with a sterile pestle. b The samples were then centrifuged for 1 min at 750 × g in a microcentrifuge. Five hundred microliters of the supernatant was transferred to a 0.45-μ cellulose acetate filter column f and centrifuged for 2 min at 10,000 × g in a microcentrifuge or until the filter was either empty or plugged.
Virus isolation
Two hundred microliters of filtered tissue homogenate was inoculated on a subconfluent culture of Vero (African green monkey kidney) cells in 1 well of a 24-well plate. Two hundred microliters of a 1:10 dilution of the same filtrate was inoculated on a second well. Cultures were incubated at 37°C for 1 hr with agitation every 15 min, and then 1 ml of medium was added to each well. Cells were observed microscopically for cytopathic effect (CPE) once per day. Supernatant from wells that had CPE was frozen at −70°C. After 5 days, plates were frozen and thawed 3 times, and 200 μl of medium from remaining wells was passed onto fresh Vero cells in 24-well plates. After similar observation, medium from wells showing CPE was frozen, and after 5 days, negative wells were discarded.
Sequence of primers and probes used in real-time reverse transcription polymerase chain reaction v1.1.
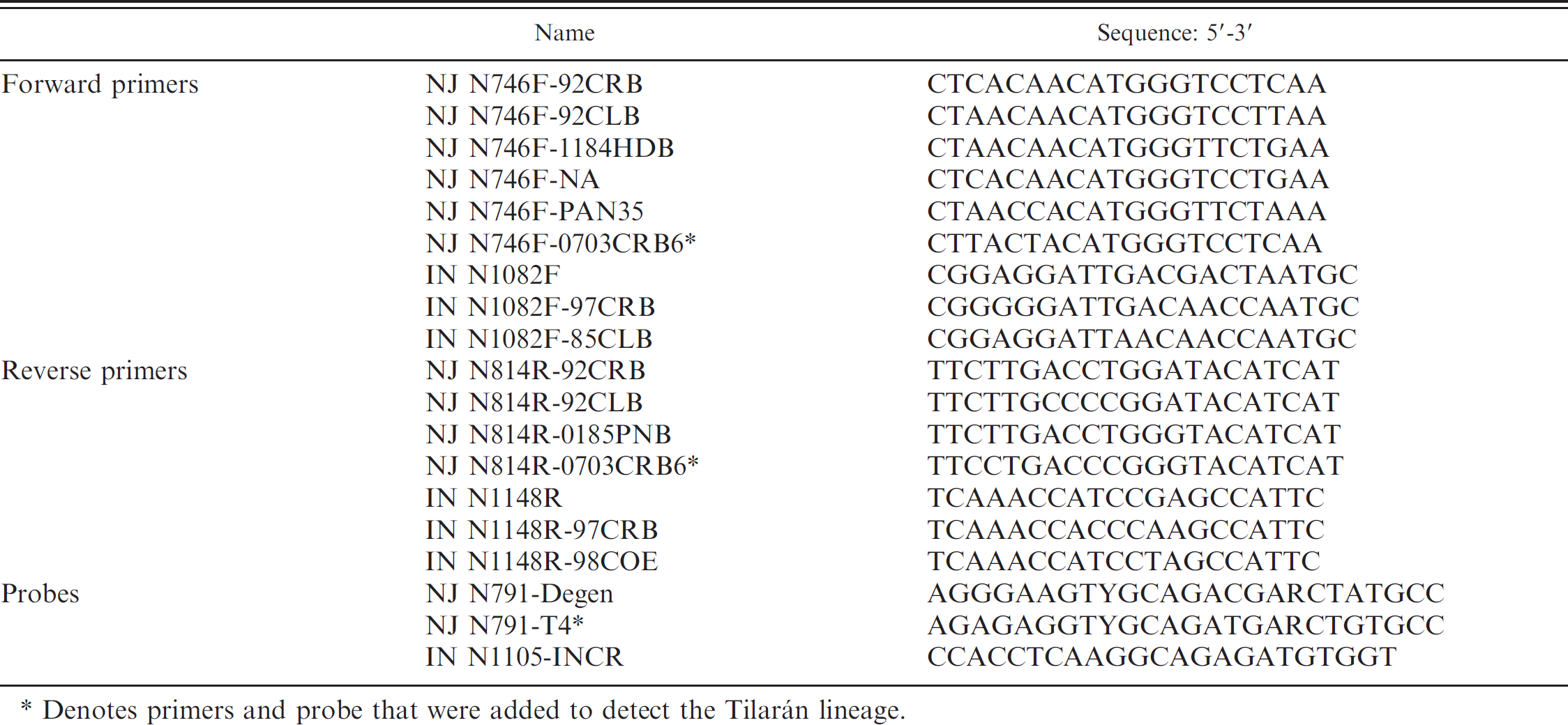
Denotes primers and probe that were added to detect the Tilarán lineage.
Immunocytochemistry
Immunocytochemistry was used to demonstrate that virus isolates were VS virus and to determine their serotype in samples tested in the Costa Rica and Panama laboratories. For this purpose, Vero cells were grown to confluence in 96-well cell culture plates, inoculated with 10 μl of a 1:100 or 1:200 dilution of supernatant from CPE-positive virus isolation samples, and incubated overnight to allow the virus to replicate. Control wells were inoculated with stock viruses or uninfected medium. Plates were inverted to empty wells, blotted, and fixed for 15 min at room temperature (approximately 28°C) with 200 μl per well of 10% neutral buffered formalin. Wells were then washed 3 times with PBS, blocked for 30 min at room temperature with 200 μl of 2% powdered milk in PBS, and washed 3 times with PBS. Two wells of each sample were incubated for 60 min with 35 μl of a 1:1,000 dilution of VSNJV-immune mouse ascitic fluid g (VR-1239) and 2 wells with 35 μl of a 1:1,000 dilution of VSINV-immune mouse ascitic fluid g (VR-1238). Wells were washed 4 times with PBS containing 2.5 ml/l Brij-35 h and incubated for 20 min with 20 μl of biotinylated multilink. i Wells were washed 4 times with PBS/Brij-35 and incubated for 20 min with alkaline phosphatase–conjugated streptavidin label. i Wells were washed 4 times with PBS/Brij-35 and developed with fast red chromogen/substrate. i Reactions were quenched with water and scored for color using an inverted microscope.
Real-time RT-PCR
A multiplex real-time RT-PCR was designed to detect the VS virus nucleocapsid gene (N), the most conserved gene and most abundant viral transcript in VS virus–infected tissues. 4,20 This test was designed to detect VS virus serotypes New Jersey (VSNJV) and Indiana (VSINV) from a broad geographic and genetic spectrum. Primers and probes were designed using Primer Express j based on nucleotide alignments of VS virus strains representing the main phylogenetic groups of VSNJV and VSINV from diverse geographic regions in the Americas. 16 Because of the virus's high genetic diversity, a single region with complete nucleotide conservation of 50 nucleotides or more within the N gene of each serotype could not be found. Therefore, the authors took the approach of designing multiple lineage-specific primers and probes targeting the same highly conserved nucleotide sequence of N. Virus-specific components consisted of 5 forward and 3 reverse primers d for VSNJV, 3 forward and 3 reverse primers d for VSINV, 1 tetrachloro-6-carboxyfluorescein (TET)–labeled probe j for VSNJV, and 1 fluorophore 6-carboxyfluorescein–labeled probe j for VSINV (Table 1). Individual primers represented genetic variability found in reference viruses representing each of the major phylogenetic groups of each serotype used in the test design. These representative strains are listed in Table 1. The targeted N gene region was between nucleotides 745 and 791 for VSNJV and 1082 and 1105 for VSINV.
The real-time RT-PCR was run in a real-time thermocyclerkl using lyophilized VSNJV and VSINV RT-PCR assay reagents. Tubes cycled through 1 reverse transcription step of 1,500 sec at 55°C, 1 denaturation step of 120 sec at 95°C, and 40 cycles of 95°C for 10 sec and 55°C for 60 sec. Following the discovery that VSNJV viruses from the Tilarán area were not detected, 2 additional primers d and 1 additional TET-labeled probe j were added. A conventional nested RT-PCR targeting the N gene was used to verify the presence or absence of VS virus from samples. 5
Diagnostic sensitivity, specificity, and accuracy of Vesicular stomatitis virus real-time reverse transcription polymerase chain reaction v1.0 tests on samples from Central America and Mexico using 2 alternative cutoff threshold cycle (Ct) values.
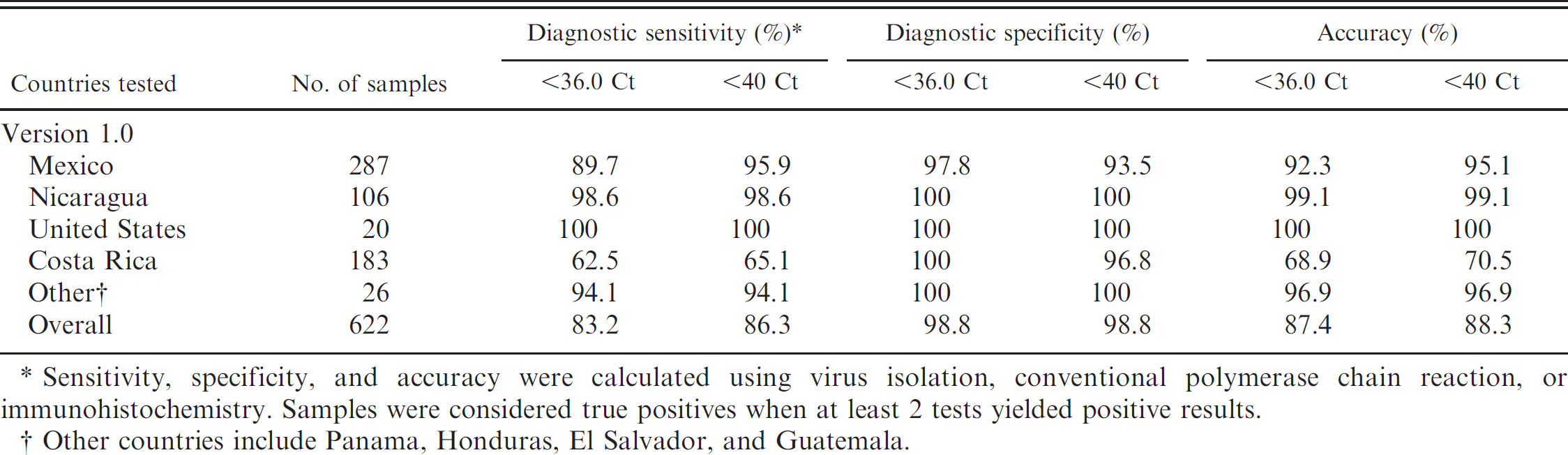
Sensitivity, specificity, and accuracy were calculated using virus isolation, conventional polymerase chain reaction, or immunohistochemistry. Samples were considered true positives when at least 2 tests yielded positive results.
Other countries include Panama, Honduras, El Salvador, and Guatemala.
DNA sequencing and analysis
DNA sequencing was performed to confirm that amplicons were actually VS viruses as previously described. 15 Briefly, reverse transcription was performed using random hexamers d and a commercially available reverse transcription kit j following the manufacturer's instructions. A portion of the VS virus phosphoprotein was PCR amplified using previously described primers. 19 Products were purified directly from the RT-PCR reaction using a commercial PCR purification kit. c The PCR products were sequenced by dideoxy sequencing using a commercial sequencing kit j on an automated sequencer. j Sequencher software version 4.1 1 was used to analyze the chromatograms. Alignments were performed using ClustalX. 23 Samples were identified as VS virus by comparison with known VS virus sequences in GenBank.
Diagnostic data analysis
Diagnostic data analyses were conducted following the World Organization for Animal Health guidelines. 12 All samples were tested by real-time RT-PCR and virus isolation. True sample status was determined by confirmatory tests including immunocytochemistry, nested PCR, and DNA sequencing of PCR products. Samples were considered true positive only when positive by real-time RT-PCR or virus isolation and at least 1 of the confirmatory tests. True-negative samples were those negative by real-time RT-PCR and virus isolation or positive by 1 of these tests but negative by 2 confirmatory tests. Diagnostic sensitivity was calculated as true positive/(true positive + false negative). 12 Diagnostic specificity was calculated as true negative/(true negative + false positive). 12 The diagnostic sensitivity and specificity of the real-time RT-PCR test were determined for 2 cutoff threshold cycle (Ct) values 36 and 40, as shown in Table 2.
Results
To thoroughly evaluate the VSNJV/VSINV multiplex real-time RT-PCR assay in the field, samples were collected over a 2-year period from Costa Rica, El Salvador, Guatemala, Honduras, Mexico, Nicaragua, and Panama. The assay was evaluated in a field-deployable format using lyophilized reagents at the Universidad Nacional School of Veterinary Medicine, at LADIVES, and at the EADC regional office.
At all locations, samples were tested by real-time RT-PCR and virus isolation. A cytopathic agent was isolated from approximately half (51.4%)ofthe samples, usually without the need for a second passage. Of 622 samples analyzed, 403 were identified as VSNJV and only 56 as VSINV. A total of 163 tested negative to VS virus. The small number of VSINV samples reflects the low occurrence of this serotype. No further distinction between serotype is made for the purposes of data analysis during this field evaluation.
Overall results by country of origin are shown in Table 2 using alternative cutoff values of 36 and 40 Ct, respectively. Using a cutoff value of <36 Ct, overall clinical sensitivity and specificity of the real-time RT-PCR v1.0 were 83% and 99%, respectively. Increasing the cutoff to <40 Ct increased the sensitivity to 86%, but there was a small decrease in specificity, particularly for the Indiana serotype. Therefore, <36 Ct was chosen as the cutoff value for this test. Interestingly, the lowest sensitivity was observed in samples originating from Costa Rica. This low sensitivity was caused by a group of samples from Costa Rica that was detected by conventional PCR or virus isolation but was not detected by the VS virus real-time RT-PCR v 1.0. These viruses showed a discrete distribution in northwestern Costa Rica centered near the Tilarán region (Fig. 1). Fifty samples from that area were partially sequenced in the nucleocapsid and phosphoprotein regions to delineate the geographic extent of the virus-positive samples that were not detected by real-time RT-PCR. Thirty-eight of these viral isolates were identical, and the remaining 12 were closely related (Fig. 2). Phylogenetic analyses demonstrated that this group of isolates represented a unique new VS virus lineage that was distinct from other viral lineages from Costa Rica but was related to a virus isolated in Honduras in 1986 (Fig. 2). Sequence comparison of the probe region of the real-time RT-PCR v1.0 with those from the Tilarán group demonstrated 6 mismatches (Fig. 3). Additional mismatches were observed in the forward and reverse primers (data not shown). Using the new sequence data from these isolates, 1 forward and 1 reverse primer, and 1 additional probe, were added to the v1.0 test (Fig. 3). With this new formulation (designated v1.1), the Tilarán group viruses were easily detectable. The v1.0 sensitivity for the detection of viral RNA in epithelial samples from Costa Rica was 62% due to the lack of detection of the Tilarán lineage. With the addition of the new primers and probe in v1.1, the sensitivity and specificity for Costa Rica samples were 93.5% and 100%, respectively, with a cutoff value of <36 Ct.

Geographic distribution of clinical samples within Costa Rica. Each dot represents a sample tested by virus isolation (VI) and the v1.0 real-time reverse transcription polymerase chain reaction (real-time RT-PCR) test. Positive samples by VI that were negative by real-time RT-PCR showed a discrete distribution in the northwestern region of the country near Tilarán.
The level of detection using v1.1 for the reference strains remained unchanged as all genetic lineages were adequately detected with the new formulation (Table 3). The final formulation of the real-time RT-PCR test v1.1 consisted of 6 forward and 4 reverse primers and 2 probes for specific detection of VSNJV (Table 1). For VSINV, there is less genetic diversity, and 3 forward and 3 reverse primers and 1 probe are sufficient to detect all viral lineages to date (Table 1).
Discussion
Vesicular stomatitis caused by VSNJV and VSINV is a common disease of cattle, swine, and horses in Central America and Mexico. Viral RNA sequencing and molecular epidemiology studies have shown that outbreaks occurring sporadically every 8–10 years in the southwestern United States represent incursions of distinct viruses originating from endemic regions, 8,15,17 although the exact mechanisms of introduction and spread remain unknown. Discrimination between VS and the clinically identical FMD, which does not exist in North or Central America, requires reliable laboratory diagnosis. Appropriate action to limit the spread of either disease results in national and international quarantines and requires that the diagnosis be made as quickly as possible.
Sequence analysis of viruses causing VS in Central and North America confirmed that, as had been shown in laboratory studies, VS virus nucleotide sequence is highly variable. This high variation makes it difficult to identify the necessary conserved nucleotide regions in all field isolates in order to design a comprehensive real-time RT-PCR. Extensive laboratory research has shown that VS virus exists as a quasi-species, in which most individuals have unique sequence variations and populations can shift rapidly. 1,3,7,14,22 Previous research has shown high genetic diversity of VS virus in endemic regions of Central America and Mexico, but remarkably within the same region, genetic sequences remain stable over time. 9 Nevertheless, independent VS virus genetic lineages exist in endemic areas delineated by specific ecological conditions. 18 Despite various efforts and extensive sequencing, no genomic region of VS virus was found that was completely conserved among viruses from the wide geographic regions where VS virus is found. Therefore, the approach of targeting the most conserved viral gene (N) and adding primers and probes targeting the sequence diversity in the same genomic region as needed to detect all viral strains was used. Despite the anticipated problems, the real-time RT-PCR based on all available Central American VS virus sequences identified all VS virus isolates tested until a new genetic lineage was found during this field validation project. The inability of the real-time RT-PCR to identify isolates from the Tilaraán region of Costa Rica led to a modification that expanded the range of the test to include the previously unknown genetic lineage. The final assay identified both VSNJV and VSINV, performed equally well on samples from cattle, horses, and swine, and identified virtually all isolates from 5 Central American countries, Mexico, and the United States. New genetic lineages might occur in the future requiring the addition or substitution of primers or probes to the current test Although this might seem cumbersome, it is expected of any RNA detection assay for a highly variable virus Data showed that the addition of near-identical primers and probes targeting the same nucleotide positions in the viral genome is a feasible approach to address this complex viral diversity problem.
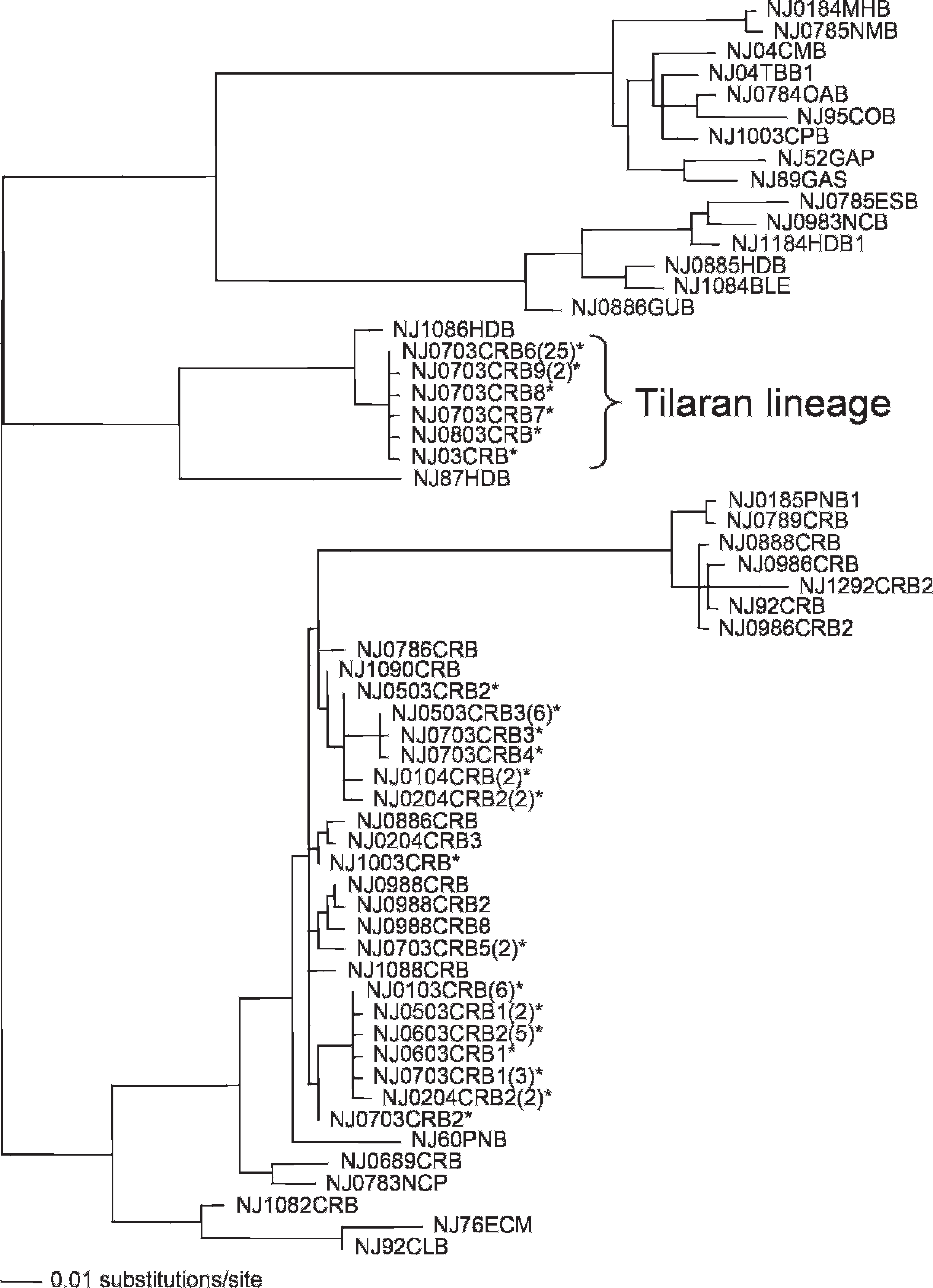
Evolutionary tree reconstructed by maximum-likelihood analysis of the P gene hypervariable region sequences from 59 viruses representing the geographic and phylogenetic breadth of Vesicular stomatitis New Jersey virus. Sequence names indicate serotype (NJ, New Jersey), month (if available), year of isolation, 2-letter state code in the United States or Mexico (MH, Michoacaán; NM, New Mexico; CM, Colima; TB, Tabasco; OA, Oaxaca; CO, Colorado; CP, Chiapas; GA, Georgia) or country of origin (ES, El Salvador; NC, Nicaragua; HD, Honduras; BL, Belize; GU, Guatemala; CR, Costa Rica; PN, Panama; EC, Ecuador; CL, Colombia), and 1-letter abbreviation of host species (B, bovine; P, porcine; S, sand fly; E, equine; M, mosquito); a number after the host species abbreviation but not in parentheses distinguishes samples from the same time and place; a number in parentheses indicates the total number of isolates possessing an identical hypervariable region sequence. Costa Rican isolates sequenced as part of the field validation are marked with an asterisk, and the novel Tilaraán lineage is indicated. The tree is midpoint rooted.

Nucleotide sequence of v1.0 New Jersey probe, nucleotide sequence of NJ0703CRB6 probe-area sequence, and nucleotide sequence of v1.1 New Jersey probe. Boxes in the NJ0703CRB6 probe-area sequence indicate nucleotide mismatches with the v1.0 New Jersey probe. Y = C or T; R = A or G.
Level of detection (LOD) of representative Vesicular stomatitis virus strains by real-time reverse transcription polymerase chain reaction v1.0 and v1.1.

The real-time RT-PCR was more sensitive than the gold standard of virus isolation. The sequence of amplicons from samples not containing live VS virus confirmed the presence of nonviable VS virus RNA. Possible explanations include sampling late in the disease course and inactivation of virus during shipment and storage. Although specific sampling, storage, and shipping protocols were provided, as with any veterinary diagnostic laboratory, the quality of the samples varied. In some cases, shipping delays and/or packing deficiencies resulted in some samples becoming thawed in transit to the laboratory. It is not surprising that many samples were positive by realtime RT-PCR despite the fact that they did not yield virus in cell culture. Serological confirmation that the animals were indeed infected was not possible because VS virus infects and causes disease even in animals with preexisting high levels of specific antibody. 24,25 However, the true sample status was always confirmed with at least 2 confirmatory positive tests.
The inability of the real-time RT-PCR to identify isolates from the Tilaraán area led to the identification of a VS virus variant that had not been discriminated from previous isolates. This variant was particularly interesting because it was isolated only in a contiguous focus constituting about 1% of the land mass of Central America and because other VS viruses were not isolated in the same focus. The Tilaraán lineage may represent the emergence and expansion of a unique sequence variant that is more fit than others for that particular ecological zone, therefore displacing existing viruses, a possible macro evolutionary manifestation of a phenomenon previously seen only in cell culture. 2 Alternatively, this virus may be a stable inhabitant of a unique micro environment that was just not previously detected.
It must be emphasized that the primers and probes used in this real-time RT-PCR were not designed for detection of Brazilian and Argentinean subtypes of VSINV (VSINV2 and VSINV3). 13 The product of the current research is a field-evaluated, rapid, robust, realtime RT-PCR that identifies all VS virus isolates in Central and North America in as little as 4 hr using dry reagents and portable instrumentation. The assay is self-correcting in that the isolation of VS virus from a PCR-negative case signals a previously unrecognized virus that can be sequenced to provide a rapid modification of the assay. A supplementary benefit of this research was the collection of a set of VS virus isolates representing most viral variants causing disease across the northern half of the global range of VS virus. Such collections are rare, and their value cannot be overstated.
Acknowledgements
The authors would like to acknowledge Irene Lopez, Luis Martin, and the personnel of Region VI from the Exotic Animal Disease Commission (EADC) in Mexico for collecting, storing, and processing of Mexico strains. The authors thank Rocio Cortes and Jorge Prendas for technical help at the School of Veterinary Medicine in Costa Rica, the LADIVES technical personnel in Panama, the Costa Rica Screwworm Eradication Program personnel, the numerous veterinarians and farmers from Central American countries that submitted samples to LADIVES, and the EADC veterinarians in Mexico who collected the field material. This project was funded by USDA-ARS project 1940-32000-041-00X.
Footnotes
a.
RNeasy® Mini Kit, Qiagen Inc., Valencia, CA.
b.
Kontes, Vineland, NJ.
c.
QiaShredder®, QIAquick® PCR Purification kit; Qiagen Inc., Valencia, CA.
d.
Invitrogen Corp., Carlsbad, CA.
e.
Hyclone, Logan, UT.
f.
Spin-x® 0.45-um column, Corning Inc., Corning, NY.
g.
American Type Culture Collection, Manassas, VA.
h.
Sigma-Aldrich, St. Louis, MO.
i.
BioGenex, San Ramon, CA.
j.
BigDye Terminator™ Sequencing Kit, 3730A automated sequencer; Applied Biosystems Inc., Foster City, CA.
k.
Smart Cycler® II, Cepheid Inc., Sunnyvale, CA.
l.
Gene Codes Corp., Ann Arbor, MI.