
Abstract
Two previously developed TaqMan fluorogenic probe-based 1-tube real-time reverse transcription polymerase chain reaction (real-time RT-PCR) assays (T1 and T2) were compared and validated for the detection of Equine arteritis virus (EAV) nucleic acid in equine semen and tissue culture fluid (TCF). The specificity and sensitivity of these 2 molecular-based assays were compared to traditional virus isolation (VI) in cell culture. The T1 real-time RT-PCR had a higher sensitivity (93.4%) than the T2 real-time RT-PCR (42.6%) for detection of EAV RNA in semen. However, the T1 real-time RT-PCR was less sensitive (93.4%) than the World Organization for Animal Health (OIE)-prescribed VI test (gold standard). The sensitivity of both PCR assays was high (100.0% [T1] and 95.2% [T2]) for detecting EAV RNA in TCF. In light of the discrepancy in sensitivity between either real-time RT-PCR assay and VI, semen that is negative for EAV nucleic acid by real-time RT-PCR that is from an EAV-seropositive stallion should be confirmed free of virus by VI. Similarly, the presence of EAV in TCF samples that are VI-positive but real-time RT-PCR-negative should be confirmed in a 1-way neutralization test using anti-EAV equine serum or by fluorescent antibody test using monoclonal antibodies to EAV. If the viral isolate is not identified as EAV, such samples should be tested for other equine viral pathogens. The results of this study underscore the importance of comparative evaluation and validation of real-time RT-PCR assays prior to their recommended use in a diagnostic setting for the detection and identification of specific infectious agents.
Keywords
Introduction
Equine arteritis virus (EAV) is an enveloped, positive-stranded RNA virus in the order Nidovirales, family Arteriviridae, and genus Arterivirus. 17 It is the causative agent of equine viral arteritis (EVA), a contagious disease of horses and other equid species. 21,22 Serologic surveys have confirmed EAV infection in equine populations in North and South America, Europe, Australia, Africa, and Asia. 14,23,26,29,30,37,46 However, the seroprevalence of EAV infection of horses varies between countries and different breeds within countries. 14,46 The majority cases of primary EAV infection are asymptomatic. 5
Occasionally infected animals will develop clinical signs of disease. The most common clinical manifestations of EVA include anorexia, depression, fever, and dependent limb edema, and less frequently, ocular and nasal discharge and conjunctivitis. Equine arteritis virus infection may also result in abortion in pregnant mares and interstitial pneumonia or pneumoenteritis in very young foals. 16,46 A variable percentage (up to 10–70%) of acutely infected stallions can become persistently infected and continue to shed virus in their semen. 46,48 Carrier stallions are the natural reservoir of EAV; they ensure the virus is maintained in equine populations between breeding seasons. 46 Furthermore, the long-term carrier state in the stallion enables the generation of genetic heterogeneity within the virus that helps to distinguish field strains of EAV. 2,3,28 The 2 principal modes of transmission of EAV are horizontal, by direct contact with infectious respiratory tract secretions from acutely infected horses, and venereal through natural breeding or artificial insemination with infective semen from persistently infected stallions. 1,5,6,47 The continued growth in international trade in horses and semen has served as a significant means of dissemination of EAV strains around the world. 1,29,46–48 Identification of the carrier stallion is therefore of critical epidemiological importance in the prevention and control of EAV infection. 5,6,46
The EAV genome consists of 2 large replicase genes (open reading frames [ORFs] 1a and 1b) and partially overlapping structural genes (ORFs 2a, 2b, 3–7). 41,42 The EAV RNA is encapsidated by the nucleocapsid protein (N; encoded by ORF7) into an icosahedral core that is surrounded by an envelope containing 6 structural proteins. Two major envelope proteins of EAV, large glycosylated protein (GP5; ORF5) and nonglycosylated membrane protein (M; ORF6), form a covalently linked heterodimer in the virion. 20 In addition to these, small envelope protein E (ORF2a) and 3 minor glycoproteins named GP2, GP3, and GP4 (ORFs 2b, 3, and 4, respectively) also contribute to the viral particle formation. 42,43 The latter 3 proteins form a covalently linked heterotrimer in the virion. 41,51
A definitive diagnosis of EAV is based on isolation of EAV in cell culture, detection of viral nucleic acid by reverse transcription polymerase chain reaction (RT-PCR) assay, or by serologic means based on testing paired (acute and convalescent) serum samples. 5,6,46,52 Detection of EAV in the semen of carrier stallions is frequently determined by virus isolation (VI) in cell culture, or much less frequently, by breeding a stallion to 2 seronegative mares and testing for development of an antibody response after 28 days. 46,52 Virus isolation is currently the World Organization for Animal Health (OIE)-approved gold standard for the detection of EAV in semen and is the prescribed test for international trade. Isolates of EAV from clinical specimens, including semen, are often confirmed in a 1-way neutralization assay using polyclonal equine sera raised against the prototype Bucyrus strain of EAV or by fluorescent antibody test using monoclonal antibodies to EAV. 25,52 These methods, although of proven reliability, are time-consuming, expensive, and cumbersome. The availability of modern nucleic acid-based assays has revolutionized diagnostic testing of clinical specimens for many infectious disease agents. 4,10–12,24,31,34,35,38,50 Compared with traditional virus isolation, these assays are frequently more sensitive, less expensive, and less time-consuming. 10–12,38 Sensitive standard RT-PCR, RT-nested PCR (RT-nPCR), and real-time RT-PCR assays have been developed for the detection of EAV nucleic acid in tissue culture fluid (TCF), nasal secretions, and semen, and these assays are being increasingly used for routine diagnostic purposes. 4,18,24,39,40,44,45,50 However, further validation of most of these assays is needed before they can be fully accepted as providing equivalent reliability to VI in cell culture for the detection of EAV.
The development of 2 TaqMan fluorogenic probe-based 1-tube real-time PCR assays for the detection of EAV nucleic acid has been previously described. 4,50 The purpose of the present study was to compare the specificity and sensitivity of these specific real-time PCR assays with each other as well as with VI for the detection of EAV in semen and in TCF. The analytical sensitivity of these 2 assays was also established by using in vitro transcribed (IVT) EAV RNA containing the complete region of ORF7 and flanking portion of ORF6 and 3′ nontranslated region (3′ NTR) of the virus.
Materials and methods
Equine semen samples, viruses, and cells
Three hundred raw equine semen samples received between April 2006 and May 2007 by the OIE EAV Reference Laboratory (Maxwell H. Gluck Equine Research Center, University of Kentucky, Lexington, KY), were tested for the presence of EAV by VI and EAV nucleic acid by 2 previously referenced real-time TaqMan RT-PCR assays. 4,50 A total of 155 TCF samples were included in the study. These comprised 1 sample containing the modified live virus (MLV) vaccine strain of EAV (ARVAC), a 61 samples containing North American and European strains of EAV, 77 EAV-negative samples, and 16 TCF samples containing a variety of other equine viral pathogens. All EAV-positive TCF samples used in the present study were previously confirmed to contain EAV in a one-way serum neutralization assay using polyclonal anti-EAV equine serum or by fluorescent antibody test using monoclonal antibodies to GP5 of EAV. 25,52 These included TCF samples containing 25 archived EAV field isolates from the OIE Reference Laboratory at the Gluck Center, and TCF samples containing 36 strains of EAV collected at the Animal Health Diagnostic Center (New York College of Veterinary Medicine, Cornell University, Ithaca, NY). The TCF samples included representation of North American and European strains of EAV. 1–3,8,9,28 To determine the specificity of these 2 real-time PCR assays, TCF containing the following equine viral pathogens was included in the study: Equid herpesviruses 1–5 (EHV-1 [ATCC VR-700], b EHV-2, EHV-3 [ATCC VR-352], b EHV-4 [ATCC VR-2230], b and EHV-5 13 ); Equine rhinitis A virus (ERAV [NVSL-0600EDV8501]) c and B (ERBV [NVSL-0610EDV85010]) c ; Equine adenovirus 1 (EAdV-1 [NVSL-001EDV8401]) c and 2 (EAdV-2); Equine influenza virus type Al (equine-l/Prague/1/56 [H7N7]; ATCC VR-297) b and A2 (equine/Miamil/63/ [H3N8; NVSL-060IDV0501], c equine/Kentucky/81 [H3N8; NVSL-040IDV0001], c equine/Alaska/29759/91 [H3N8; NVSL-020IDV9101]) c ; and Salem virus, a novel paramyxovirus of horses. 27
The low passage RK-13 cell line (ATCC CCL37; passage level 194–204) b and high passage RK-13 cell line (RK-13 KY; passage level 399–409) were maintained in Eagle minimum essential medium (EMEM) d supplemented with 10% ferritin-supplemented bovine calf serum, e 1% penicillin and streptomycin, d and 0.1% amphotericin B (1,000 μg/ml). d The overlay medium used for inoculated cultures was of 0.75% carboxymethyl cellulose (CMC) f in supplemented EMEM.
Virus isolation
Isolation of EAV from equine semen samples was attempted in both high and low passage RK-13 cell lines according to the standard laboratory protocol used by the OIE Reference Laboratory. 52 Briefly, semen samples were sonicated for 45 sec (3 × 15 sec), and sperm and cellular debris were sedimented by centrifugation (2,800 × g, 10 min) at 4°C. Serial decimal dilutions (10-1-10-3) of the supernatant of each sample were made in supplemented EMEM and 1 ml of each dilution was inoculated into each of 2 × 25-cm2 flasks containing confluent monolayers of RK-13 cells. Flasks were incubated at 37°C for 1 hr before being overlaid with supplemented EMEM containing 0.75% CMC. Flasks were incubated at 37°C and checked for the appearance of cytopathic effect (CPE) on post-inoculation days 3 and 4. If there was no detectable CPE. a second blind passage was performed on day 4. The RK-13 cell monolayers were fixed and stained with a 1% crystal violet solution containing 1% formaldehyde on postinoculation day 5 for the first passage and postinoculation day 4 for the second passage in cell culture.
RNA isolation
Viral nucleic acid was directly isolated from semen and TCF samples using a commercial kit. g , 8 Briefly, semen or TCF samples were microcentrifuged at 13,800 × g for 2 min, and 140 μl of supernatant was removed and used for nucleic acid extraction according to the manufacturer's instructions. The viral nucleic acid was eluted in 60 μl of nuclease-free water and stored at −80°C.
Generation of in vitro transcribed RNA
The analytical sensitivity of the 2 real-time TaqMan RT-PCR assays was determined using a decimal dilution series (101–1010 molecules/reaction) of in vitro transcribed (IVT) EAV RNA containing complete ORF7 and the flanking regions of ORF6 and 3′ NTR of the EAV genome. Briefly, a 636-bp fragment of EAV (nucleotide [nt] 12069–12704: numbered according to GenBank accession number Y07862 49 ) was PCR amplified from the plasmid containing the complete genomic sequence of the virulent Bucyrus strain of EAV (pEAVrVBS) 7 using primers 12069P ([12069–12088] 5′TTTGTTATAGTTGGAAGAGC3′) and 12704N ([12681–12704] 5′GGTTCCTGGGTGGCTAATAACTA C3′), and cloned into the pDrive cloning vector according to the manufacturer's instructions. h The plasmids were purified using a commercial kit, i and the authenticity and orientation of the insert was determined by sequencing both strands of DNA with T7 and SP6 reverse and forward primers. Following sequencing, the recombinant plasmid with ORF7 and flanking ORF6 and 3′ NTR sequence downstream of the SP6 promoter was named pORF7.SP6 and used to generate IVT ORF7 RNA. Runoff pORF7.SP6 RNA transcripts were generated from Bam HI-linearized pORF7.SP6 plasmid according to a previously described protocol. 8 Briefly, a 50-μl reaction volume containing 2 μg of Bam HI-linearized pORF7.SP6 plasmid DNA; 2.5 μl of RNAguard RNase inhibitor (37 U/μl) j ; 5 μl of m 7 G(5′)PPP(5′)G RNA cap structure analogue k ; 5 μl of ribonucleotides ATP (adenosine 5′-triphosphate), CTP (cytidine 5′-triphosphate), GTP (guanosine 5′-triphosphate), and UTP (uridine 5′-triphosphate; 10 mM each mix)1; 2.5 μl of 100 mM dithiothreitol (DTT) m ; 2.5 μl of SP6 RNA polymerase m ; 5 μl of 1 mg/ml acetylated BSA m ; and 1 × transcription buffer m was incubated at 37°C for 2 hr. The template plasmid DNA was then digested with RNase-free DNase I. n The RNA transcripts were purified using a commercial column, o eluted in 50 μl of water, and quantified by spectrophotometrical analysis. The IVT ORF7 RNA was stored at −80°C until used. The concentration of the IVT ORF7 RNA molecules per microliter was calculated according to the following formula:

The dilution of RNA transcripts (101–1010 molecules) was carried out in RNase- and DNase-free molecular biologic grade water containing 0.1 mg/ml acetylated BSA m and 0.74 U/μl RNAguard RNase inhibitor. j
Real-time TaqMan reverse transcription polymerase chain reaction assays
A 1-tube real-time TaqMan RT-PCR assay was performed using the TaqMan One-Step RT-PCR Master Mix p in a 7500 Fast Real-Time PCR System. q The primers and probes used in the 2 assays were identical to those previously used for each respective assay (Table 1). 4,50 Every sample was tested in duplicate in each assay. Briefly. 25 μl of RT-PCR mixture for each reaction contained 12.5 μl of 2 × Master Mix without UNG (uracil-N-glycosylase), 40 × MultiScribe and RNase Inhibitor Mix. 900 nM of forward and reverse primers (0.45 μl), 250 nM probe (0.625 μl), nuclease-free water (5.35 μl), and 5 μl of test sample RNA. The following thermocycling conditions were used under standard mode as per manufacturer's recommendation: 30 min at 48°C, 10 min at 95°C, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. Each RT-PCR run included a control without RNA (containing the reaction mix with 5 μl of water [no template control]) and positive controls containing IVT ORF7 RNA. The analytical sensitivity of each real-time PCR assay was determined using IVT ORF7 RNA.
Statistical analysis
Statistical evaluation of the performance of semen and TCF-based real-time PCR assays for the detection of EAV nucleic acids was carried out to estimate and compare the sensitivity, specificity, accuracy, and reproducibility of the T1 and T2 real-time PCR assays in relation to VI. For the purpose of the analysis, T+ denotes a positive real-time PCR test result and 1+ denotes a VI-positive specimen, with T- and I- denoting a negative real-time PCR test result and a VI-negative specimen, respectively. Accordingly, the sensitivity (Se) of either diagnostic test is the likelihood that an EAV carrier stallion tests positive, namely Se = Pr(T+|I+), and its specificity (Sp) is Sp = Pr(T-|I-). Accuracy is defined as the likelihood of a correct test result; this is a composite measure of diagnostic performance based on both sensitivity and specificity. Test repeatability was characterized by the coefficients of variation when duplicates of serial decimal molecule dilutions (101–1010) of IVT ORF7 RNA were used in 5 independent assays.
Primers and probes used in the T1 and T2 real-time reverse transcription polymerase chain reaction (RT-PCR) assays.
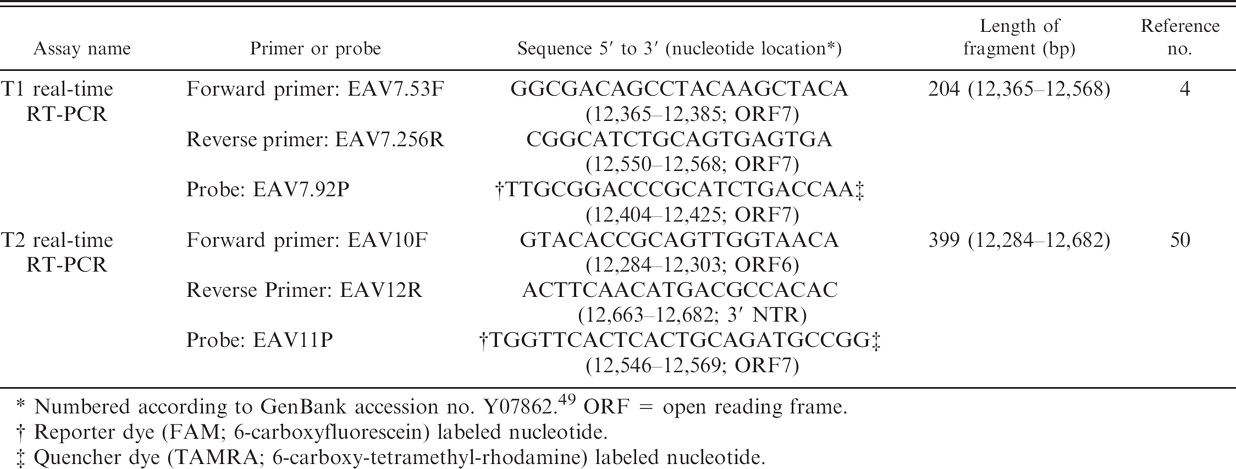
Numbered according to GenBank accession no. Y07862 49 ORF = open reading frame.
Reporter dye (FAM; 6-carboxyfluorescein) labeled nucleotide.
Quencher dye (TAMRA; 6-carboxy-tetramethyl-rhodamine) labeled nucleotide.
Sensitivity, specificity, and accuracy were calculated using Clopper-Pearson 95% exact binomial confidence intervals. 19 Exact binomial methods were also used to test the hypothesis of equal sensitivity (i.e., that Se1 = Se2) versus the two-sided alternative for the semen and tissue culture fluid PCRs. Empirical coefficients of variation (CV) were calculated as the sample standard deviation divided by the sample mean. Statistical significance was set at α = 0.05. The analysis was performed using the S-Plus 8.0 r and Minitab 15 s software packages.
Results
Analytical sensitivity of the two real-time reverse transcription polymerase chain reaction assays
In order to determine the analytical sensitivity of the 2 real-time PCR assays in detecting EAV nucleic acid, serial decimal molecule dilutions (101–1010) of IVT ORF7 RNA containing EAV ORF7 with flanking regions of ORF6 and 3′ UTR were tested in duplicate. The assays were repeated 5 times, each time with freshly diluted IVT ORF7 RNA. The IVT ORF7 RNA molecule numbers were calculated based on the molecular weight and concentration of the IVT RNA. Regression analysis confirmed linearity in both assays (T1: R 2 = 0.9984; T2: R 2 = 0.9979; Fig. 1). The T1 real-time PCR was linear over the full molecule range tested (101–1010) with high reproducibility and a mean CV ranging from a low value of 0.32% to a high of 3.18%. While the T2 real-time PCR was also linear, the analytical sensitivity of this test was only 102 molecules of IVT RNA. The range of nine orders of magnitude (102–1010 molecules) detected by the T2 real-time PCR was reproducible with a mean CV from a low value of 1.39% to a high of 9.55%. The T1 real-time PCR always had a lower cycle threshold (Ct) value than the T2 real-time PCR over all the IVT RNA dilutions tested. These data indicate that the T1 real-time PCR has a higher analytical sensitivity than the T2 real-time PCR.
Evaluation of the two real-time reverse transcription polymerase chain reaction assays for detection of Equine arteritis virus in tissue culture fluid samples
Nucleic acids extracted from a total of 155 TCF samples were tested by these 2 real-time PCR assays. Of the TCF samples positive for infectious EAV, 62 (62/62) and 59 (59/62) were positive by the T1 real-time PCR and T2 real-time PCR, respectively (Table 2). There is no significant difference between the sensitivity of both assays when RNA from TCF containing EAV was evaluated (P = 0.248). Sensitivity of the T1 real-time PCR for detecting EAV nucleic acid in TCF was 100.0% (95% confidence interval [CI]: 95.3–100.0%) whereas the sensitivity of the T2 real-time PCR was 95.2% (95% CI: 86.5–99.0%; Table 3). There was no detectable fluorescence signal in the non-template controls containing molecular biologic grade water nor in the tubes that contained nucleic acid from other viral pathogens, confirming that both assays were highly specific (100%) for the detection of EAV nucleic acid. In terms of accuracy, the T1 real-time PCR correctly diagnosed 155 out of 155 samples, providing an accuracy of 100.0% (exact 95% CI: 98.1–100.0%). In the case of the T2 real-time PCR, it correctly diagnosed 152 out of 155 samples, providing an accuracy of 98.1% (exact 95% CI: 94.4–99.6%).
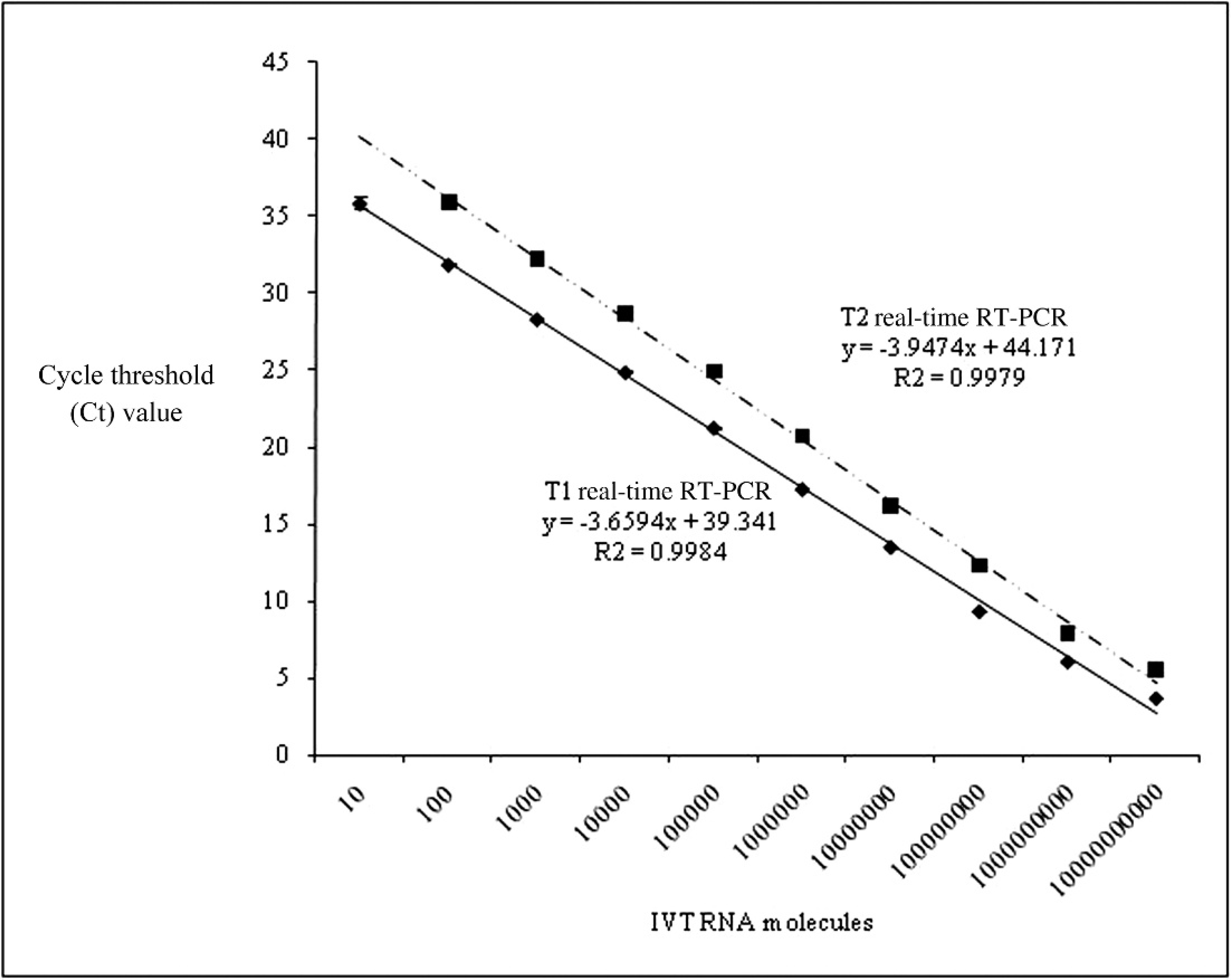
Comparison of analytical sensitivity between the 2 real-time reverse transcription polymerase chain reaction assays (RT-PCR), T1 (y = −3.6594x + 39.341, R 2 = 0.9984) and T2 (y = −3.9474x + 44.171, R 2 = 0.9979), using in vitro transcribed open reading frame 7 RNA.
Evaluation of the two real-time reverse transcription polymerase chain reaction assays for the detection of Equine arteritis virus nucleic acid in semen samples
Of the 300 equine semen samples tested, 61 (61/300) were positive for EAV by VI isolation in the RK-13 cell lines (Table 2). When nucleic acid extracted from these semen samples was tested, 57 (57/300) and 26 (26/300) were positive for the presence of EAV RNA using the T1 real-time PCR and the T2 real-time PCR, respectively. The T2 real-time PCR assay had a significantly higher number of false negatives (35) as compared with the T1 real-time PCR assay. 4 Of the 4 false-negative semen samples detected by the T1 real-time PCR, 3 had very low infectivity titers (1 × 101 to 4.5 × 101 pfu/ml). However, 1 semen sample had a moderate infectivity titer (2.9 × 103 pfu/ml).
When these 2 real-time PCR assays were subjected to the exact binomial test of equal sensitivity, the sensitivity of the respective assays was significantly different (P < 0.001). The sensitivity of the T1 real-time PCR in detecting EAV nucleic acid in semen was 93.4% (exact 95% CI: 84.1–98.2%), whereas the sensitivity of the T2 real-time PCR was much lower (42.6%; exact 95% CI: 30.0–55.9%; Table 4). There was no detectable fluorescence signal in nontemplate controls containing molecular biologic grade water nor in the tubes containing RNA from the VI-negative semen samples, confirming that both assays were highly specific (100%) for the detection of EAV nucleic acid. Since the T1 real-time PCR correctly diagnosed 296 out of 300 samples, the accuracy of this assay was 98.7% (exact 95% CI: 96.6–99.6%), compared with the T2 real-time PCR, which correctly diagnosed 265 out of 300 samples for an accuracy of 88.3% (exact 95% CI: 84.1–91.7%).
Detection of Equine arteritis virus (EAV) nucleic acid in semen and tissue culture fluid using the T1 and T2 real-time reverse transcription polymerase chain reaction (RT-PCR) assays.
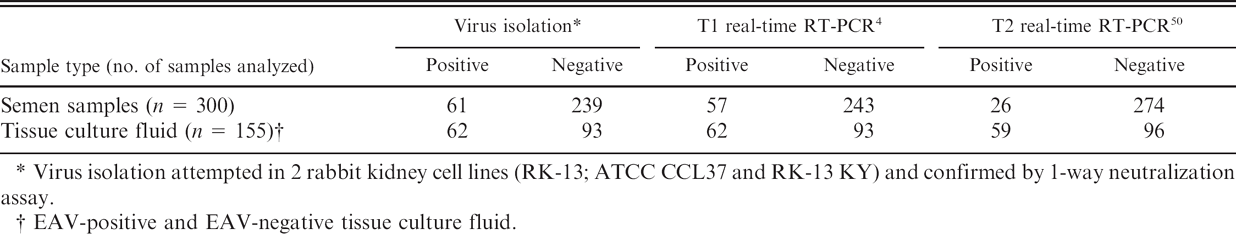
Virus isolation attempted in 2 rabbit kidney cell lines (RK-13; ATCC CCL37 and RK-13 KY) and confirmed by 1-way neutralization assay.
EAV-positive and EAV-negative tissue culture fluid.
Sensitivity, specificity, and accuracy of 2 real-time reverse transcription polymerase chain reaction (RT-PCR) assays for detection of Equine arteritis virus in tissue culture fluid as compared with virus isolation.
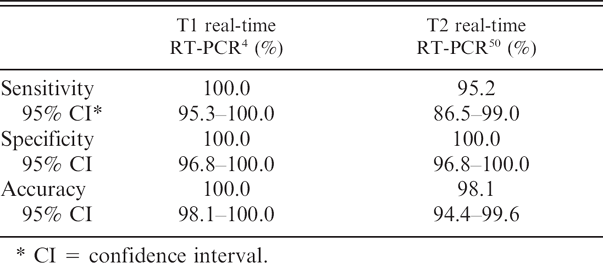
CI = confidence interval.
Discussion
The 1-tube real-time PCR assay for EAV provides a simple, rapid, and reliable method for the detection and identification viral nucleic acid in equine TCF and semen. The real-time PCR has the following important advantages over the standard 2-step RT-PCR: 1) eliminating the possibility of cross-contamination between samples with previously amplified products since the sample tube is never opened, and 2) reducing the chance of false-positive reactions because the real-time PCR product is detected with a sequence specific probe. The original reports describing the respective real-time PCR assays to detect EAV nucleic acid suffered from the limitation that each study was based on testing only a small number of samples (21 TCF and 20 semen samples in the T1 study 4 ; 28 TCF and 33 semen samples in the T2 study 50 ). Clearly, before recommending either real-time PCR assay as an alternative to VI for the detection of EAV in TCF and semen, each assay needed to be more comprehensively validated using a significantly larger number of specimens.
Sensitivity, specificity, and accuracy of 2 real-time reverse transcription polymerase chain reaction (RT-PCR) assays for detection of Equine arteritis virus in semen as compared with virus isolation.
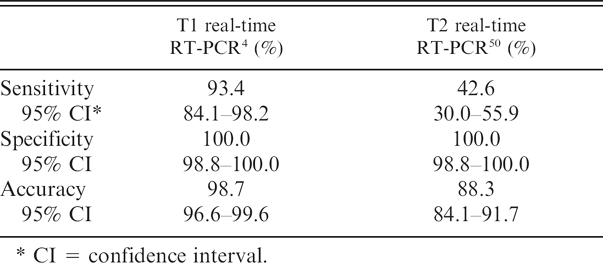
CI = confidence interval.
In the present study, the analytical sensitivity of the T1 real-time PCR and T2 real-time PCR assays were first evaluated using IVT ORF7 RNA. Both T1 real-time PCR and T2 real-time PCR assays were shown to have a very high analytical sensitivity for IVT ORF7 RNA, detecting 10 and 100 RNA molecules, respectively. The T1 real-time PCR was at least 10 times more sensitive than the T2 real-time PCR and consistently had a lower Ct value with different IVT RNA concentrations. The difference in analytical sensitivity between these 2 assays is likely attributable to the size of the amplicons they generated. The T1 real-time PCR amplicon length was much shorter than the T2 real-time PCR amplicon (204 bp vs. 399 bp), which suggested that it was amplified more efficiently than the longer real-time PCR product. Furthermore, it has been reported that shorter PCR products are more responsive to reaction conditions, and allow primers and probe to bind to the target molecule more efficiently. 15
The primary goal of this study was to compare the diagnostic sensitivity and specificity of 2 published real-time PCR assays for the detection of EAV nucleic acid based on testing an extensive number of TCF and semen samples. With the exception of the primers and probe, all of the other test conditions were identical for both assays, which were performed in parallel at the same time. Under the conditions of this study, the T1 real-time PCR has a higher diagnostic sensitivity than the T2 real-time PCR in detecting EAV nucleic acid in semen (93.4% vs. 42.6%) and to a lesser degree in TCF (100.0% vs. 95.2%) using the TaqMan One-Step RT-PCR Master Mix Reagents kit. p
Several factors could have adversely affected the diagnostic sensitivity of the T2 real-time PCR assay, including the length of the real-time PCR product previously mentioned, nucleotide mismatch in the primer and probe binding regions, and the use of proprietary reagents and cycle parameters that are optimized for use with the ABI 7500 Fast Real-Time PCR system. q The authors aligned 54 EAV sequences available in GenBank (nt # 12069 to 12645 [ORF6–ORF7]; data not shown) to determine the degree of conservation in the regions to which the primers and probes are directed in the 2 assays. The forward primer-binding region (nt # 12365–12385) of the T1 real-time PCR was highly conserved (no mismatches) among the aligned EAV strains. However, both reverse primer- (nt # 12550–12568) and probe- (nt # 12404–12425) binding regions had 1 nucleotide mismatch. The reverse primer-binding region (nt # 12663–12682) of the T2 real-time PCR was highly conserved (only 17 aligned sequences cover the 3′ NTR). In contrast, the forward primer- (nt # 12284–12303) and probe- (nt # 12546–12569) binding regions had 3 and 5 nucleotide mismatches, respectively. This clearly suggests that with respect to field strains of EAV, the forward primer- and/or probe-binding regions targeted by the T2 real-time PCR is less conserved than the regions targeted by the T1 real-time PCR. It has been shown that a single nucleotide mismatch in the primer- or probe-binding site may have minimal or zero effect on PCR amplification or detection. 32,33 In contrast, 3 or 4 nucleotide mismatches at certain critical locations within the primer- or probe-binding region can have a considerable effect including complete failure of detection. 32,33 In light of this, the mismatches of the forward primer and probe within the T2 real-time PCR assay may have significantly reduced the efficiency of the PCR reaction, especially in the case of specimens containing low quantities of EAV nucleic acid. In order to compare the 2 assays in a standard and normalized manner, a commercial kit and manufacturer's recommended real-time RT-PCR cycle parameters were used in this study. Therefore, the reagents and real-time RT-PCR cycle conditions used in this study were not identical to the original T2 assay. These modifications may also have contributed to the reduced sensitivity of the T2 assay.
There was good overall agreement between the T1 real-time PCR and the VI assay, which is the current gold standard for the detection of EAV in semen. 52 However, under the conditions of this study, the T1 real-time PCR was less sensitive (93.4%) than the VI assay for detection of EAV nucleic acid in semen. The sensitivity of the real-time PCR assay versus VI may be adversely affected by various factors including use of the one-step RT-PCR method, reaction conditions (e.g., Mg2+ concentration, annealing temperature), amount of virus in the original sample, RT-PCR inhibitory substances in certain samples, mutations in the primer- and probe-binding regions of the template (see above), inefficient RNA extraction from semen samples, and the potential for RNA degradation before testing. The T1 real-time PCR assay was designed to balance both sensitivity and ease of use so that the procedure could be performed rapidly and on a large scale. A single-step real-time PCR was performed with a commercial kit and RT-PCR conditions pre-optimized by the manufacturer, notwithstanding the fact that the 1-step method is reported to be less sensitive than a 2-step RT-PCR procedure. 36 The T1 real-time PCR allows detection of EAV in clinical samples or TCF containing at least 50 viral RNA molecules per ml when 140-μl specimen is used for RNA extraction. 4 The titer of EAV in semen from carrier stallions can vary from 1 × 101 pfu/ml to >105 pfu/ml. Over 97% of the semen samples submitted to the OIE EAV Reference Laboratory contain at least 1.5 × 102 pfu/ml and the remaining samples contain <1.5 × 102 pfu/ml. Three of the 4 false-negative semen samples in the T1 real-time PCR had very low infectivity titers ranging from 1 × 101 pfu/ml to 4.5 × 101 pfu/ml. Interestingly, all these semen samples were from stallions in the process of spontaneously clearing the carrier state. Notwithstanding their very low infectivity content, it was possible to isolate EAV in RK-13 cells from all 3 semen samples. The fourth false-negative semen sample was of poor quality in that a large amount of smegma-like material was present, which may contain some unknown real-time PCR inhibitors. This clearly indicates that the T1 real-time PCR as described is less sensitive than VI attempted in RK-13 cells. In view of this finding, any semen sample negative in the T1 real-time PCR assay that is from an EAV-seropositive stallion should also be screened for virus in RK-13 cells.
It should be noted, however, that genomic variation among field isolates of EAV may reduce the accuracy of the real-time PCR, even when the primers and probe are based on the most conserved region of EAV genome (ORF7). Therefore, the presence of EAV in VI-positive but real-time PCR-negative TCF samples should be confirmed in a 1-way neutralization assay using an EAV-positive antiserum or by fluorescent antibody test using monoclonal antibodies to GP5 of EAV. 25,52 If negative for EAV, such TCF samples should be tested for other equine viral pathogens.
In summary, 2 previously described real-time PCR assays were compared with each other, as well as with attempted VI in cell culture for the detection of EAV in TCF samples and semen. The T1 real-time PCR had a higher sensitivity than the T2 real-time PCR in detecting EAV nucleic acid in TCF and semen samples. Neither real-time PCR was superior to the OIE-prescribed VI test (gold standard) for the detection of EAV in semen. In light of these findings, semen from an EAV-seropositive stallion that is negative for viral nucleic acid by real-time PCR should also be tested by VI. The findings of this study illustrate the importance of comparative evaluation and validation of real-time PCR assays prior to their recommended use in a diagnostic laboratory for the detection and identification of infectious agents.
Acknowledgements
This study was supported by funds from American Quarter Horse Association and the Kentucky Agricultural Experiment Station, College of Agriculture, University of Kentucky. Dr. Zhengchun Lu is the recipient of a Geoffrey C. Hughes Foundation graduate fellowship. EHV-2 and EHV-5 were kindly provided by the late Dr. William H. McCollum, Gluck Equine Research Center, University of Kentucky, and by Dr. Stephanie Bell, University of California, Davis, respectively. Equine adenovirus 2 isolated from a foal with pneumonia was kindly provided by Mr. Stephen Sells, Livestock Disease Diagnostic Center. University of Kentucky.
Footnotes
a.
EAV modified live vaccine (ARVAC), Fort Dodge Animal Health, Fort Dodge, IA.
b.
Equid herpesvirus-1, −3, and −4, Equine influenza virus type Al, and low passage RK-13 cell line, American Type Culture Collection, Manassas, VA.
c.
Equine rhinitis A virus and B, Equine adenovirus 1, and Equine influenza virus type A2, National Veterinary Services Laboratory, Ames, IA.
d.
Eagle minimum essential medium, penicillin and streptomycin, and amphotericin B, Mediatech, Inc., Herndon, VA.
e.
Ferritin-supplemented bovine calf serum, Hyclone Laboratories, Inc., Logan, UT.
f.
Carboxymethyl cellulose, Sigma-Aldrich, St. Louis, MO.
g.
QIAamp Viral RNA isolation kit, Qiagen, Inc., Santa Clara. CA.
h.
Qiagen PCR cloning kit, Qiagen, Inc., Santa Clara, CA.
i.
QIAamp Miniprep kit, Qiagen, Inc., Santa Clara, CA.
j.
RNAguard RNase inhibitor, Amersham Bioscience, Pittsburgh, PA.
k.
m7G(5′)PPP(5′)G RNA cap structure analogue, New England BioLabs, Beverly, MA.
l.
10 mM rATP, rCTP, rGTP, and rUTP, Amersham Bio-science, Pittsburgh, PA.
m.
100 mM DTT, SP6 RNA polymerase, 1 × transcription buffer and acetylated BSA, Promega Corp., Madison, WI.
n.
RNase-free DNase I, Ambion, Austin, TX.
o.
QIAamp RNeasy Mini kit, Qiagen Inc., Santa Clara, CA.
p.
TaqMan One-Step RT-PCR Master Mix, Applied Biosystems, Foster City, CA.
q.
7500 Fast Real-Time PCR System, Applied Biosystems. Foster City, CA.
r.
S-Plus 8.0, Insightful Corp., Seattle, WA.
s.
Minitab 15, Minitab, Inc., State College, PA.