
Abstract
Initial results demonstrating the feasibility of a multiplexed liquid array immunoassay for foot-and-mouth disease viral antigen detection and simultaneous serotype differentiation are presented. Serotype-specific antibodies from rabbit and guinea pig hyperimmunesera were isolated and prepared for use in a multiplexed, bead-based assay. The performance of all of the available antibodies as both capture and detector reagents was evaluated in the multiplexed system to establish a combination exhibiting the highest homotypic responses and lowest heterotypic reactions. The multiplexed assay was evaluated against inactivated cell culture supernatant samples of the same subtype as the virus used to raise the capture and detector antibodies. Distinct serotype differentiation was observed, except in the case of serotype SAT1. Subsequently, cell culture supernatant samples from a larger pool of viral subtypes were analyzed. Distinct serotype differentiation was obtained when analyzing cell culture supernatant samples from viral serotypes C, Asia, and SAT3, irrespective of the subtype. However, limitations of the current antibody pairs were realized in some inconclusive results obtained when analyzing samples from a broader range of O, A, and SAT2 subtypes. The results obtained in this initial study will be used to further optimize the assay using polyvalent or monoclonal antibodies and move toward the analysis of clinical samples.
Luminex™ liquid array technology enables the simultaneous evaluation of a sample with multiple detection reagents (multiplexing) in a single assay. There are many examples of the creative use of this technology 12 for the detection of biothreat agents 13,14 and other infectious disease. 10,11,15 Multiplexed analysis of a sample with numerous detection reagents gives increased confidence in assay results and eliminates well-to-well variation sometimes observed when comparing multiple single-well, singleplex assays. Multiplexing cuts down on reagent and consumable consumption, thereby reducing cost. In addition, significant reductions in labor costs and an expanded capability to deal with surge capacity are also possible when running multiplexed assays in single wells. Here, initial results demonstrating the feasibility of a liquid array, multiplexed immunoassay for foot-and-mouth disease (FMD) viral antigen detection, and serotype differentiation are presented.
Foot-and-mouth disease virus (FMDV) is a member of the picornaviridae family. It consists of a small, unenve-loped capsid and a single strand of positive sense RNA. There are 7 serotypes of the virus C, O, A, Asia, SAT1, SAT2, and SAT3 and many subtypes. FMD diagnosis and serotyping is most commonly carried out using an epithelial suspension but is also conducted with blood, saliva, and vesicular fluid using an enzyme-linked immunosorbent assay (ELISA), 4,7,16 which enables a positive sample to be confirmed in a few hours. However, if the virus concentration is very low or inconclusive ELISA results are obtained, then cell culture is used to amplify the virus for analysis. If the multiplicity of infection is low, then it can take 2 to 4 days for a cytopathic effect to become apparent before subsequent cell culture supernatant samples are analyzed for the final diagnosis. All assays currently used for FMDV identification and serotyping require multiple assays, where each assay tests for a single serotype and the results are analyzed and interpreted as a composite to identify the serotype.
The multiplexed assay described here was based on a combination of guinea-pig (GP) and rabbit (Ra) FMD serotype-specific polyclonal antibodies, isolated from antisera using affinity columns. The multiplexed assay was assessed for its performance in the serotype differentiation of inactivated 6 viral cell culture supernatant samples of 19 different FMD viral subtypes.
All reagent dilutions and assays were carried out using filtered (0.22 μm) PBS-TBN (phosphate-buffered saline, pH 7.4; Tween 20 0.02% vol/vol; bovine serum albumin [BSA] 0.1% wt/vol; NaN3 0.02% wt/vol). Ra 9 and GP 5 antisera were produced as described previously. Antisera were prepared against specific subtypes: C1 Noville, O1BFS 1860, A24 Cruzeiro, Asia 1 Shamir, SAT1 Ken 4/98, SAT2 Zim 5/81, and SAT3 Zim 4/81. Antisera samples (5 ml) were filtered using Steriflip-GP filters (0.22 μm). a Subsequently, antibodies were isolated from the antisera samples using Montage Antibody Purification Pro-sep A spin columns a per kit instructions. Resulting antibody solutions were desalted and transferred into ∼ 1.5-ml PBS/ 0.02% wt/vol NaN3 using Amicon Ultra 15 centrifugal filter units molecular weight cut off (MWCO) 30 kDa a per kit instructions and stored at 4°C. Recovered protein yields ranged from 6 μg/ml (GP SAT3) to 25 μg/ml (Ra SAT1) determined by UV absorbance at 280 nm (Nano-drop spectrophotometer). b One milliliter (1.25 × 107) of carboxylate beads c was used as received and coated with the polyclonal antibodies as described previously 13,15 using 0.5 ml antibody solution (125 μg/ml in 0.1 M 2[N-morpholino] ethane sulfonic acid buffer, pH 4.5). To determine successful antibody coupling, 2-μl beads were diluted into 98 μl PBS-TBN. Two microliters of anti-rabbit IgG (whole molecule)-R-phycoerythrin antibody produced in goat d and R-phycoerythrin AffiniPure F(ab′)2 fragment donkey anti-guinea pig IgG (H+L) e were added to the Ra antibody-coated beads and GP antibody-coated beads, respectively, and incubated in the dark for 20 minutes. The samples were directly analyzed in a Bio-Plex f to 10,000 events per bead class with a 50-μl sample size to determine the median fluorescent intensity (MFI). Successful coating procedures yielded MFIs of 9,000 to 19,000 for Ra antibody-coated beads and 17,000 to 21,000 for GP antibody-coated beads. A set of 4 controls (instrument control [IC], fluorescent control [FC], antibody control [AC], and negative control [NC]) built into the bead mixture monitors and reports every step of the assay. 14 The details and concentrations at which the control beads are coated have been described previously. 13,15 The 10X bead mixtures were formulated in PBS-TBN to a final concentration of ∼7 × 105 of each bead class/ml. Following formulation, 10 μl of the 10X bead mixture diluted in 90 μl PBS-TBN was analyzed in duplicate in a Bio-Plex f to 10,000 events per bead class with a 50-μl sample size to determine if the bead counts in each class were approximately equal. If the bead count of a particular class was significantly (>30%) lower than the others, a compensatory amount of that bead was added to the bead mixture. The bead mixtures were stored at 4°C in the dark and diluted 10-fold directly before use. For the biotinylated detector antibody cocktail (b-Abc), typically, 2 ml of each antibody solution was prepared at 1 mg/ml in PBS/0.02% wt/vol NaN3. Two milligrams of EZ-Link sulfo-NHS-LC-biotin g was freshly prepared in 364 μl deionized (DI) water and 40.5 μl was added to each antibody solution and gently agitated for 30 minutes. Volumes and amounts were adjusted if a different volume/concentration of antibody solution was biotinylated. Excess, unreacted biotin was removed using 5-ml Zeba Desalt spin columns g per kit instructions. The b-Abc was prepared at a working concentration as a mixture of the pertinent biotinylated Ra and/or GP antibodies, each at 3 μg/ml and biotin-SP conjugated Affinipure rabbit anti-chicken IgY (IgG), Fc fragment specific e as a control at 0.02 μg/ml in PBS-TBN. Reporter reagent, streptavidin-R-phycoerythrin (SA-PE), h was prepared in PBS-TBN at 24 μg/ml and diluted 10-fold with PBS-TBN directly before use for a working concentration of 2.4 μg/ml. Viral cell culture supernatant samples were prepared 8 and BEI-inactivated 2 as described previously. Supernatant samples were stored at −80°C and were thawed and diluted in PBS-TBN directly before use where appropriate.
Assays were carried out as described previously. 15 In brief, 100 μl of sample was incubated with 50 μl bead mixture for 20 minutes. Samples were washed twice and resuspended in 100 μl PBS-TBN and incubated with 50 μl b-Abc for 15 minutes. Samples were washed and resuspended in 100 μl PBS-TBN and incubated with 50 μl SA-PE for 5 minutes. The samples were washed and resuspended in 100 μl PBS-TBN. Finally, the suspended beads were transferred to a round-bottomed 96-well plate i for analysis with a Bio-Plex f configured to count a minimum of 100 beads per class and a 50-μl sample size.
Figure 1 depicts the basis of liquid array technology for multiplexed immunoassay-based serotype differentiation. Serotype-specific capture antibodies are each covalently attached to a unique bead class. These antibodies capture the specific antigen, and detection is via a cocktail of biotinylated serotype-specific antibodies and the subsequent addition of a reporter molecule, SA-PE. The complex is analyzed in a flow cytometer, and the MFI of the reporter is used to quantify the amount of antigen captured on each serotype-specific antibody-coated bead. The ELISA routinely used by the world reference laboratory (WRL) for FMD for antigen detection and serotype differentiation is based on serotype-specific Ra antisera as a capture agent and GP serotype-specific antisera as a detector reagent. 3,4 For this initial development of a multiplexed serotyping immunoassay, 7 serotype-specific polyclonal Ra and 7 serotype-specific polyclonal GP antibodies were isolated from antisera and were evaluated against cell culture supernatant samples from the 7 major serotypes and various subtypes of FMDV. Each antibody was employed as both a capture and as a detector antibody, and all possible combinations were evaluated. With consideration given to homotypic and heterotypic reactions with each antibody as a capture and detector reagent, a set of capture-detector antibodies exhibiting the highest homotypic and lowest heterotypic reactions was then selected for further analysis. The intensity of the response and the serotype differentiation for SAT1 was unfortunately extremely low with the antibody pairs available, and therefore SAT1 was eliminated from this further evaluation. Therefore, the bead mixture comprised capture antibodies GP C; Ra O, GP A, GP Asia, GP SAT2, and GP SAT3 plus controls IC, FC, AC, and NC. The b-Abc combination was GP C, Ra O, GP A, GP Asia, Ra SAT2, and GP SAT3 plus control. This combination was used to analyze undiluted viral cell culture supernatant samples obtained from the same viral subtype as the capture and detector antibodies, and the results are shown in Figs. 2a-2f. From comparison of the absolute MFI values on beads C, O, A, and Asia samples (Figs. 2a-2d, respectively), homotypic reactions were significantly stronger (more than double) than heterotypic reactions. Results for SAT2 and SAT3 samples (Figs. 2e and 2f, respectively) are not as distinct, with lower intensity responses and greater heterotypic reactions. However, while the SAT2 and SAT3 assays were not as clearly differential as the others, the assay still provided a conclusive result. The responses from the control beads were monitored throughout all experiments (data not shown) and remained consistent across all experiments, demonstrating consistency in assay procedure; a consistently low negative control indicated a lack of nonspecific binding in the complex sample matrix.
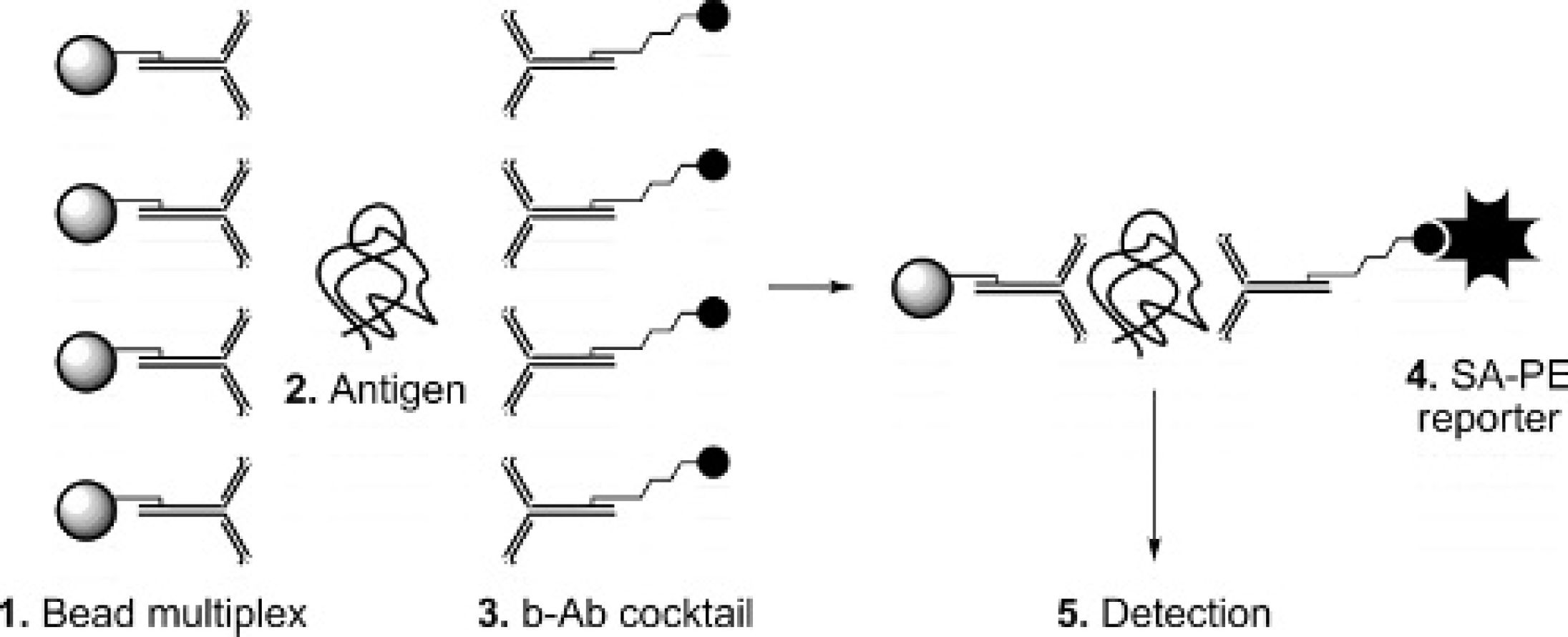
Representation of a liquid array serotyping immunoassay.
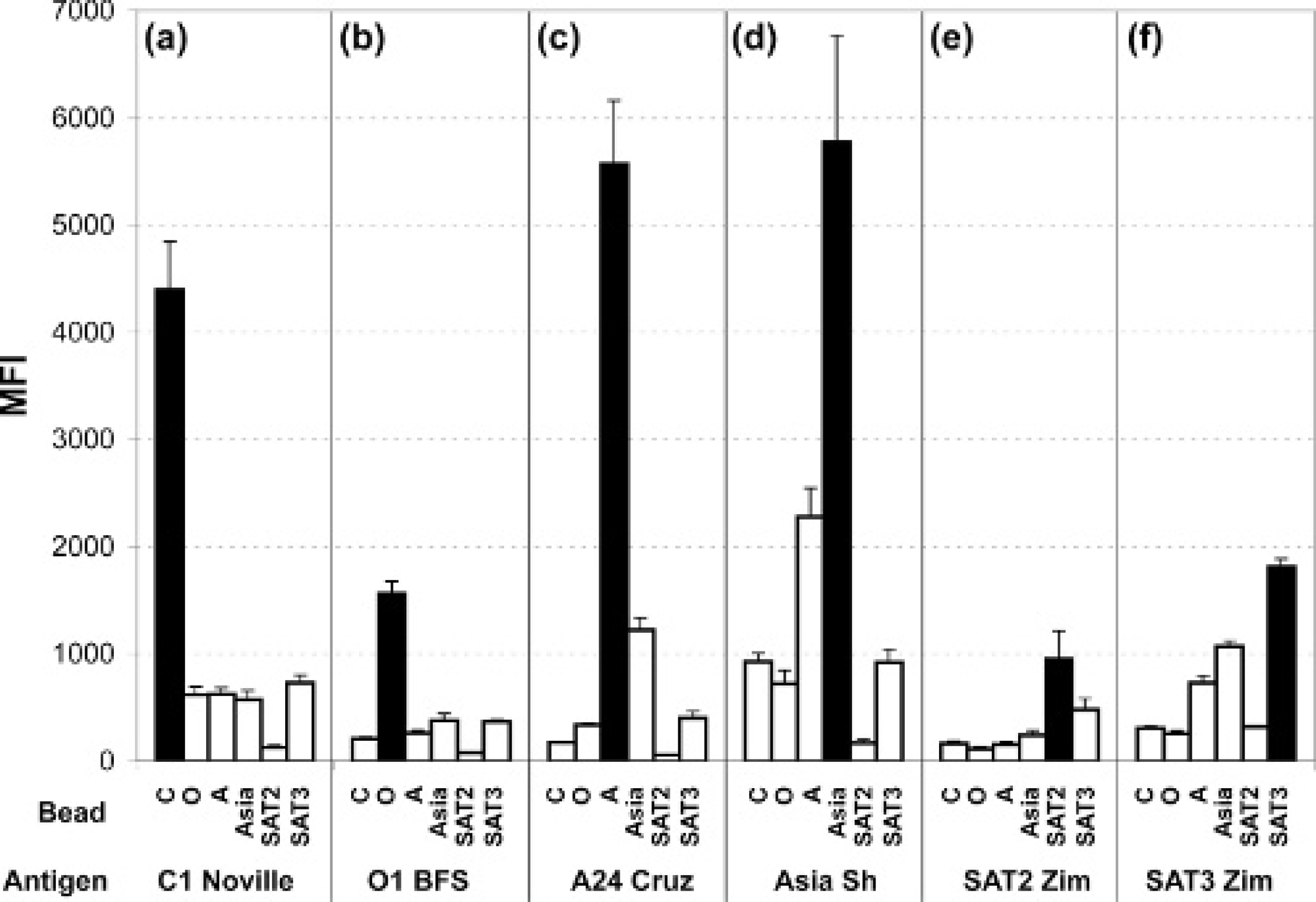
Performance of multiplexed liquid array serotyping assay. Bead multiplex: GP C; Ra O, GP A, GP Asia, GP SAT2, and GP SAT3 plus controls IC, FC, AC, and NC (control data not shown); b-Abc GP C, Ra O, GP A, GP Asia, Ra SAT2, GP SAT3. Median fluorescence intensity (MFI; y-axis) values from each serotype-specific antibody-coated bead when assayed against undiluted viral cell culture supernatant. Sample serotype and subtype (left to right):
To assess the performance of the multiplexed assay across subtypes, 12 additional cell culture supernatants from a variety of viral subtypes were analyzed. Table 1 lists the MFI with standard deviation on each serotype-specific antibody-coated bead (left to right) in the multiplex when analyzed against buffer blank and undiluted cell culture supernatant samples from the different subtypes (top to bottom); the results depicted graphically in Figure 2 are included for comparison. Horizontal comparison of MFI values on each bead obtained for a particular sample gives a comparison of the magnitude of the homotypic and heterotypic reaction in each case. The assay showed distinct serotyping across subtypes for C, Asia, SAT3, and all but 1 sample (A ARG/87) for A. Unfortunately, the O- and SAT2-coated bead showed inaccurate serotyping with the additional subtypes analyzed with significant heterotypic reactions. These results highlight some limitations of the multiplexed serotyping immunoassay when using this particular set of polyclonal antibody pairs. However, the use of polyvalent antibodies 1 for broader subtype reactivity or serotype-specific monoclonal antibodies on beads and a universal detector reagent 3 to minimize heterotypic reactions would likely significantly improve multiplexed assay sensitivity and specificity. The multiplexed serotyping assay is convenient to execute with 55-minute assay preparation and 45-minute reading and is conducive to automation. Therefore, further capture and detector reagent optimization will likely result in greater serotype differentiation, and with multiple assays carried out in a single well, liquid array technology provides a convenient, high-throughput platform.
Median fluorescence intensity (MFI) values on each serotype-specific antibody-coated bead in multiplexed serotyping immunoassay.
Horizontal comparison of the MFI values on each bead obtained when analyzing a particular sample gives a comparison of the relative reaction (homotypic vs. heterotypic) on each bead in the multiplexed system. Homotypic reactions are bolded.
denotes undiluted cell culture supernatant samples.
denotes the capture and detector antibodies are raised against that antigen subtype.
denotes inaccurate serotyping. Each point was performed in triplicate. Buffer blank is an average of 9 repeats in each of the 19 different experiments (i.e., 171 repeats).
Acknowledgements. The authors thank the Institute for Animal Health, Pirbright, UK, for providing viral isolates for cell culture supernatant sample preparation and Timothy C. Doyle for assistance in preparation of figures. This work was carried out under the auspices of the US Department of Energy by the University of California, Lawrence Livermore National Laboratory under Contract W-7405-Eng-48 with funding (JP, JIO, BJH, and MTM) from the US Department of Homeland Security Agricultural Domestic Demonstration and Application Program. This work (AC, TJS, HJH) was also supported through funds of the Laboratories Directorate of the Canadian Food Inspection Agency and the Chemical, Biological, Radiological and Nuclear Research and Technology Initiative (CRTI) No. 0196RD.
Footnotes
a.
Millipore Corp., Billerica, MA.
b.
Nanodrop Technologies, Inc., Wilmington, DE.
c.
Luminex Corp., Austin, TX.
d.
Sigma Co., St. Louis, MO.
e.
Jackson ImmunoResearch Laboratories, West Grove, PA.
f.
Bio-Rad Laboratories, Hercules, CA.
g.
Pierce Biotechnology Inc., Rockford, IL.
h.
Caltag Laboratories, Invitrogen, Carlsbad, CA.
i.
Corning Inc., Acton, MA.