
Abstract
The aims of the current study were to identify Mycoplasma suis antigens and develop a multiplex microbead immunoassay (MIA). A M. suis–expression library was screened for immunogens using sera from infected pigs. Based on bioinformatics, putative antigens were identified within positive inserts; gene fragments were expressed and purified as polyhistidine fusion proteins, and immunoreactivity was confirmed by Western blot. Selected antigens were used to develop a MIA. Sera from noninfected and infected pigs were used to set the median fluorescent intensity (MFI) cutoffs and as positive controls, respectively. Assay specificity was tested using sera from pigs seropositive for other pathogens (2 different pigs seropositive for each pathogen). Samples from 51 field pigs and 2 pigs during the course of acute (pig 1) and chronic (pig 2) infections were tested using MIA, indirect hemagglutination assay (IHA), and quantitative polymerase chain reaction (qPCR). Sixteen reactive plaques (52 genes) were detected. A heat-shock protein (GrpE), a nicotinamide adenine dinucleotide–dependent glyceraldehyde 3-phosphate dehydrogenase (GAPN), and 4 proteins from paralogous gene families (PGFs) were identified as antigens by Western blot. While GrpE, GAPN, and 1 PGF protein were strong antigens, the others were not suitable as MIA targets. A MIA using GrpE, GAPN, and the strongly reactive PGF protein was developed. Cross-reactivity with sera from pigs infected with Mycoplasma hyopneumoniae, Porcine circovirus-2, Porcine parvovirus, Porcine reproductive and respiratory syndrome virus, and Porcine respiratory coronavirus with this MIA was not observed. Pig 2 was consistently positive by MIA and qPCR, whereas pig 1, initially negative, seroconverted before becoming qPCR positive. Only 2 samples (from pig 1) were IHA positive. Five (9.8%) field samples were qPCR positive and 40 (78.43%) were positive for all 3 MIA antigens; however, all were IHA negative. In summary, the MIA is specific and more sensitive than qPCR and IHA, providing simultaneous evaluation of antibody response to M. suis antigens.
Introduction
Mycoplasma suis infection is highly endemic in pig herds. While acute infection by this red blood cell pathogen can lead to life-threatening hemolytic anemia, clinical signs associated with chronic infection may be subtle or even absent. 16 The chronic form of infection is most commonly seen in the field. 16 However, acute disease has reemerged in farrowing units, where lactation failure and lethargic sows have been observed, and in feeder pigs, where high mortality rates have been reported.3,26
Molecular diagnostic assays, such as the quantitative polymerase chain reaction (qPCR) assay, are the current methodologies of choice for the detection of M. suis–infected animals. 5 An indirect hemagglutination assay (IHA), using a crude preparation of M. suis antigens from whole blood of infected animals, 25 is the only serologic test commercially available. Animal use issues related to the production of these crude antigens prompted the replacement of the IHA with recombinant protein immunoassays. Accordingly, 3 highly conserved bacterial proteins of M. suis have been identified as antigens, heat-shock protein DnaK, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and inorganic pyrophosphatase (Ppa),7,9,10 and individual enzyme-linked immunosorbent assays (ELISAs) have been developed using these targets.7,15,28 The recent sequencing of 2 strains of M. suis provides a rich source for identification of additional antigens.4,20 Both strains have an abundance of paralogous gene families (PGFs), dedicating over 40% of their genomes to encoding these proteins. The purpose of such large numbers of similar genes is not known, but certain mucosal mycoplasmas use such homologous sequences to generate protein variants through phase-variation or recombination. 2 These genetic mechanisms are used by the organisms to avoid host immunity and establish persistent infection. 2 However, the immunogenicity of PGF of M. suis has not yet been established. The antigenic core of this hemotrophic mycoplasma is likely much larger than what is currently known and may also include some of these PGF proteins.
Advances in bead-based flow cytometry have made multiplex microbead immunoassay (MIA) an attractive alternative to conventional methods for antibody detection (e.g., ELISA, fluorescent antibody test, and Western blot).13,27 These microsphere-based flow cytometric methodologies enable the simultaneous detection of antibodies against several antigens in the same reaction container using a small sample volume. Furthermore, serologic testing can be accomplished in a high-throughput, fully quantitative platform.13,27 The utilization of microspheres in liquid-phase conditions with bright reporter dyes also has shown higher sensitivity than conventional immunoassays. 13 It is likely that the use of MIA in veterinary medicine will be greatly expanded in the future for detection of antibodies against various antigens of the same pathogen, or different pathogens in a single sample. Therefore, the aims of the current study were to identify M. suis antigens and develop a multiplex fluorescent microbead immunoassay to detect M. suis antibodies in the serum of pigs.
Materials and methods
Serum samples
Sera from 2 pigs, produced in a previously described experiment, 5 were used in the current study (pigs 1 and 2, females, 45 days old). In summary, pig 1, a splenectomized, M. suis–infected animal, served as a model of acute disease. Pig 2 was not splenectomized, was clinically healthy, and hemotrophic mycoplasmas were not observed on peripheral blood smears at any time of the experiment. 5 However, this pig was positive for M. suis by qPCR throughout the experiment. Based on the absence of clinical signs and failure to detect organisms by light microscopy, pig 2 was considered chronically infected. 5
Pools of sera from pigs 1 and 2 served as positive controls and were made as follows. First, for the library screening and Western blot, 6 serum samples from each infected pig collected 1–3 weeks apart (pig 1: days 28, 35 [first day of a positive qPCR], 46, 72, 81, 113; pig 2: days 4, 21, 53, 72, 113, 121) were pooled, and immunoglobulin G (IgG) was purified using a commercial kit. a Purified IgG was then absorbed against Escherichia coli strain XL1-Blue antigens to remove cross-reactive antibodies. 23 Second, for MIA, using the same serum samples described above, the pig 1 sample pool was considered the acute disease control, and the pig 2 sample pool was considered the chronic disease control. To mimic the handling of field samples, these pools were neither purified for IgG nor absorbed against E. coli antigens.
As negative (nonimmune) controls, 20 serum samples from M. suis–free cesarean-derived and colostrum-deprived (CDCD) pigs were pooled. For the library screening and Western blot, IgG was purified and absorbed against E. coli antigens, whereas, for the MIA, the pooled sample was not subjected to these procedures.
Serum samples collected at various time intervals from pigs 1 and 2 were tested using MIA to follow the course of infection (pig 1: days 4, 7, 18, 21, 28, 35, 46, 71, 80, 102, 113, n = 11; pig 2: 0, 4, 21, 53, 71, 113, 121, n = 7). In addition, field serum samples from 51 (3–6-month-old) pigs submitted to the Veterinary Diagnostic Laboratory, College of Veterinary Medicine, Iowa State University (Ames, Iowa) were tested.
Whole blood DNA extraction and qPCR
Ethylenediamine tetra-acetic acid (EDTA) blood samples, collected at the same time as serum samples, were also available from all pigs. DNA extraction procedures and M. suis–specific qPCR results for pigs 1 and 2 were previously reported. 5 Field pig samples of EDTA blood were extracted using a commercial kit b and subjected to a M. suis–specific qPCR assay. 5
Mycoplasma suis DNA extraction and expression library analysis
Mycoplasma suis was separated from the blood of an experimentally infected pig at peak of bacteremia using a combination of centrifugation procedures and filtration with 0.45-µm syringe-top filter as previously described. 4 Mycoplasma suis high molecular weight DNA was extracted using a commercial kit c and purified using drop dialysis in 1× phosphate buffered saline. The isolated high molecular weight DNA was then digested to completion using the 6 base pairs cutter (GAATTC) restriction enzyme EcoRI. d The digested DNA was purified using organic extraction h and ligated/packaged into an EcoRI predigested commercial vector. e
Plaque blotting for the identification of immune reactive plaques was performed as previously described, with modifications.19,24 Briefly, the library
e
phages were transfected into E. coli strain XL1-Blue MRF’ and plated onto NZY plates.
f
Each plate was incubated at 37°C for 6–8 hr and then overlaid with isopropyl β-
Bioinformatic analyses and candidate antigen selection
Phagemid insert sequences were analyzed j against the genome of M. suis strain Illinois to identify the genome region and coding DNA sequences (CDSs). Several characteristics of protein localization and similarity were analyzed for the identification of potential immunogens, including transmembrane domains, k lipoprotein prediction, l signal peptides, m and homology to known antigens. j Coding DNA sequences or CDS fragments containing such characteristics were chosen for expression, purification, and analysis by Western blot.
Protein expression, extraction, and purification
Coding DNA sequence fragments or complete CDSs of putative immunogens within inserts were selected and cloned into a commercial vector and propagated into chemically competent E. coli. n Plasmids were purified using a commercial kit o and sequenced at Purdue Genomics Core Facility (West Lafayette, Indiana) to ensure that the insert was in-frame and in the correct orientation. Plasmids were transformed into E. coli BL21 Star (DE3), n and protein expression was later performed for 24 hr at 37°C by using lysogeny broth media with carbenicillin (50 µg/ml) and 1 mM of IPTG as an inducer. Recombinant expression was confirmed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 4–15% gel; 15 µl of the induced and noninduced culture pellets were mixed with Laemmli buffer and loaded into the gel). 14 A Western blot was performed for the identification of the polyhistidine-tag component in the expressed protein as described.17,22 Noninduced cultures and BL21 Star (DE3) cells n were used as negative controls.
Pellets obtained from the cultures by centrifugation (1,000 × g for 15 min) were subjected to protein extraction by 3 freeze–thaw cycles and 10 sonication cycles of 10 sec each in equilibration buffer (50 mM NaH2PO4, 300 mM NaCl, 6 M guanidine HCl, 10–20 mM imidazole, pH 7.4). The solution was centrifuged at 15,000 × g for 5 min, and the pellet and supernatant were subjected to SDS-PAGE to ensure successful protein extraction. Finally, purification of the protein from the supernatant was performed using cobalt purification columns p under denaturing conditions (equilibration buffer; and elution buffer: 50 mM NaH2PO4, 300 mM NaCl, 6 M guanidine HCl, 150 mM imidazole, pH 7.4).
Western blot analyses
In order to confirm the molecular size of the proteins and presence of a polyhistidine-tag, a Western blot was performed as previously described.17,22 Immunoreactivity of the recombinant proteins was also confirmed by Western blot with modifications. Briefly, recombinant proteins were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. 22 Membranes were then incubated overnight with hyperimmune anti–M. suis IgG or nonimmune IgG (0.5 mg of each; diluted in TBST 2% skim milk) and washed for 2 hr with TBST with buffer replacement every 10 min for the initial hour and subsequently every 30 min. Following washes, membranes were incubated with rabbit anti-pig horseradish peroxidase–conjugated IgG q (2 mg/ml; 1:15,000 in TBST 2% skim milk) for 1 hr and washed with TBST for 40 min with buffer replacement every 10 min. Immunoreactive bands were visualized using TMB. g
Fluorescent microbead immunoassay
Coupling of polystyrene microbeads r with different amounts of protein was accomplished as described by the manufacturer in a stepwise fashion until an optimal amount was identified. Beads were counted with a hemocytometer, and 100 beads/µl/well were used in the assays. The MIAs were performed in filter plates with a vacuum manifold. Briefly, 96-well filter plates were blocked for 2 min with 100 µl of phosphate buffered saline (pH 7.4) with 1% bovine serum albumin (PBN). Fifty microliters of microbeads in PBN were added to each well followed by 50 µl of the test sera diluted in TBST 1% bovine serum albumin. The plate was then incubated at RT for 90 min with shaking (600 rpm) followed by 3 washes with 100 µl of PBN using a vacuum manifold. Microbeads were resuspended in 50 µl of PBN and incubated with 50 µl of rabbit biotin–conjugated anti-pig IgG s (10 mg/ml; 1:200 in PBN) for 1 hr at RT with shaking. The plate was again washed twice with PBN and incubated with 50 µl of phycoerythrin-conjugated streptavidin (4 µg/ml in PBN) t for 30 min with shaking. Following 2 additional washes, microbeads were resuspended by pipetting up and down 10 times in 120 µl of PBN. The assay was analyzed (70 µl of each well) in a commercial instrument u with corresponding software, v and at least 100 beads per well were analyzed. Data was reported as MFIs. Positive controls (acute and chronic disease controls, pigs 1 and 2, respectively), a negative control (pooled sera from CDCD pigs), a background control (only secondary antibody), and all serum samples were evaluated in duplicates in every tested plate.
Optimization of the MIA
In order to determine the optimal dilution of porcine sera for use in the assay, MFIs generated from serial dilutions of positive controls (acute and chronic disease controls, pigs 1 and 2, respectively) and the negative control (pooled sera from CDCD pigs) were compared. The optimal working dilution was determined as the dilution giving the highest dynamic range (ratio between MFIs from the lowest positive control and the negative control, respectively) with the lowest background. The optimum dilution for the secondary antibody (biotin-conjugated anti-pig IgG) was similarly assessed.
The assay cutoff for a negative result was established by testing duplicates of each serum sample from the 20 CDCD pigs described above; the cutoff was determined as the mean of all 20 samples plus 3 standard deviations (SD). Intra-assay variability was assessed using 7 replicates of both positive controls (acute and chronic disease) and the negative control in the same plate. Interassay variability was performed with duplicates of each control in 3 separate runs. The coefficient of variation (CV) among MFIs was calculated as a measurement of the assay’s variability. The assay’s analytical specificity was tested by using sera from pigs seropositive for common porcine pathogens (2 sera from different pigs seropositive for each pathogen): Mycoplasma hyopneumoniae, Porcine circovirus-2 (PCV-2), Porcine parvovirus (PPV), Porcine reproductive and respiratory syndrome virus (PRRSV), and Porcine respiratory coronavirus (PRCV).
Indirect hemagglutination assay
Serum samples from pig 1, pig 2, and field animals were tested by IHA at an independent diagnostic laboratory (Veterinary Diagnostic Laboratory, College of Veterinary Medicine, University of Illinois, Urbana-Champaign, Illinois). The assay is available at that location and was performed as previously described. 25
Statistical analyses
In order to calculate the agreement between the MIA and the qPCR and IHA, a sample was classified as seropositive if positive for at least 2 out of the 3 antigens. The kappa coefficient of association was then used to evaluate such agreements. The Mann–Whitney U test was used to compare MFI values between field samples that were positive and negative by qPCR. All analyses were performed using commercial software. w A test was considered statistically significant when the P value was <0.05.
Results
Antigen identification
The titer of the unamplified EcoRI library was 3.25 × 106 plaque-forming units/ml, with 98.3% of recombinants. Following the screening of approximately 3 × 104 phages, a total of 16 positive plaques were identified, representing 52 CDSs of which 25 (48.1%) were part of 7 PGFs (Table 1). The genomic locations of each plaque are also shown in Table 1. Because mycoplasmas translate the UGA codon as tryptophan instead of a stop codon, it is likely that only fragments of proteins were expressed in the expression system. For this reason, only fragments (lacking UGA codons) of genes likely to encode immunogenic proteins based on bioinformatics were selected for further analyses, as described previously for Mycoplasma haemofelis. 17 A total of 13 protein fragments were cloned, expressed, and purified (National Center for Biotechnology Information accession nos. of DNA fragments are listed in Table 1). One of these proteins, the heat-shock protein GrpE (full-length; no UGA codons present), was not identified in the library screening, but it was chosen based on previous reports of its immunogenicity potential in other bacteria.12,18,29 Finally, Western blot analyses with sera from infected animals showed that 6 out of these 13 recombinants were immunogenic. These include GrpE (MSU_0474), GAPN (MSU_0029), and 4 hypothetical proteins from 4 different PGFs (MSU_0108, MSU_0110, MSU_0184, MSU_794; Fig. 1).
Reactive phage plaques and the characteristics of the inserts according to Mycoplasma suis strain Illinois.*
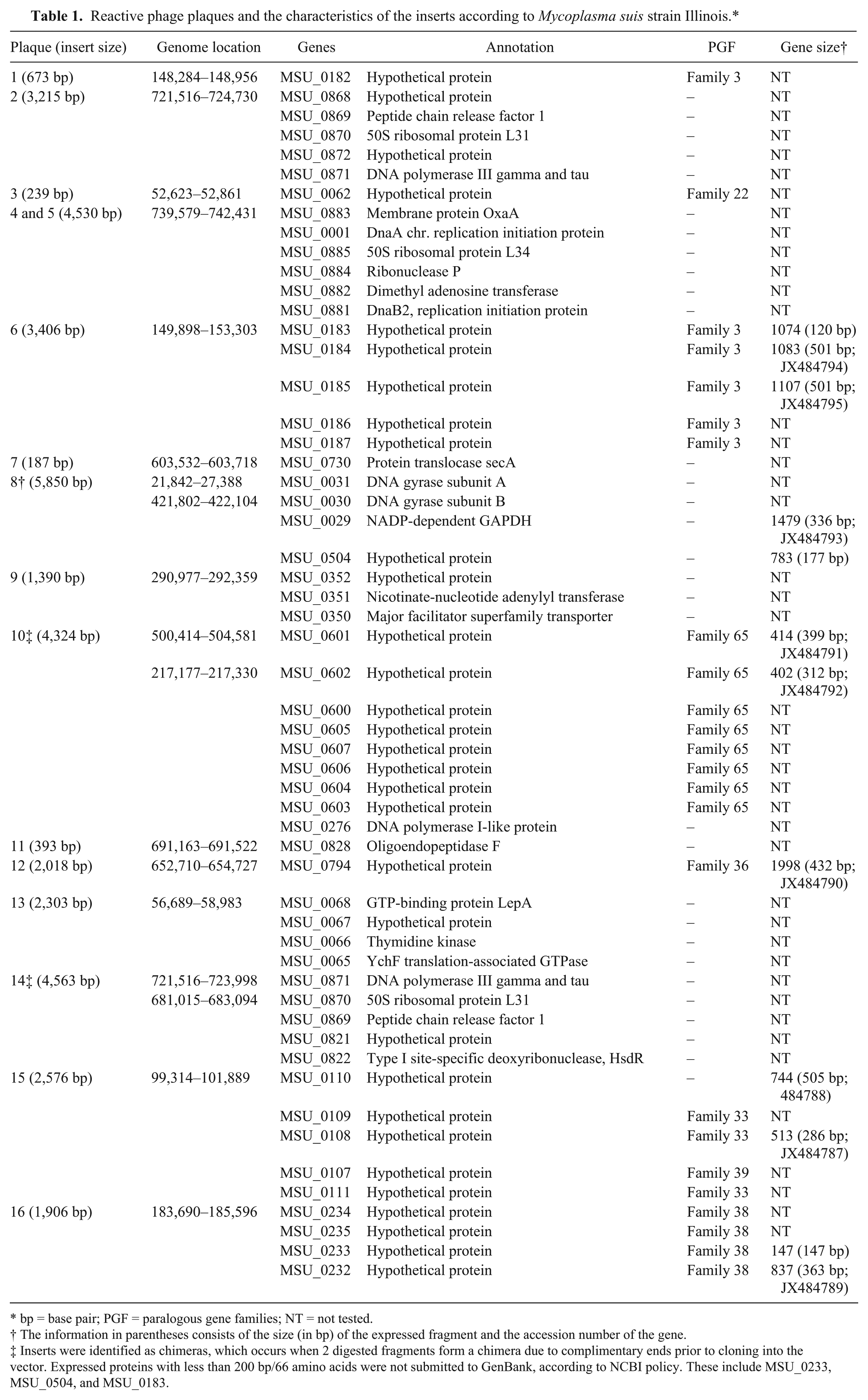
bp = base pair; PGF = paralogous gene families; NT = not tested.
The information in parentheses consists of the size (in bp) of the expressed fragment and the accession number of the gene.
Inserts were identified as chimeras, which occurs when 2 digested fragments form a chimera due to complimentary ends prior to cloning into the vector. Expressed proteins with less than 200 bp/66 amino acids were not submitted to GenBank, according to NCBI policy. These include MSU_0233, MSU_0504, and MSU_0183.
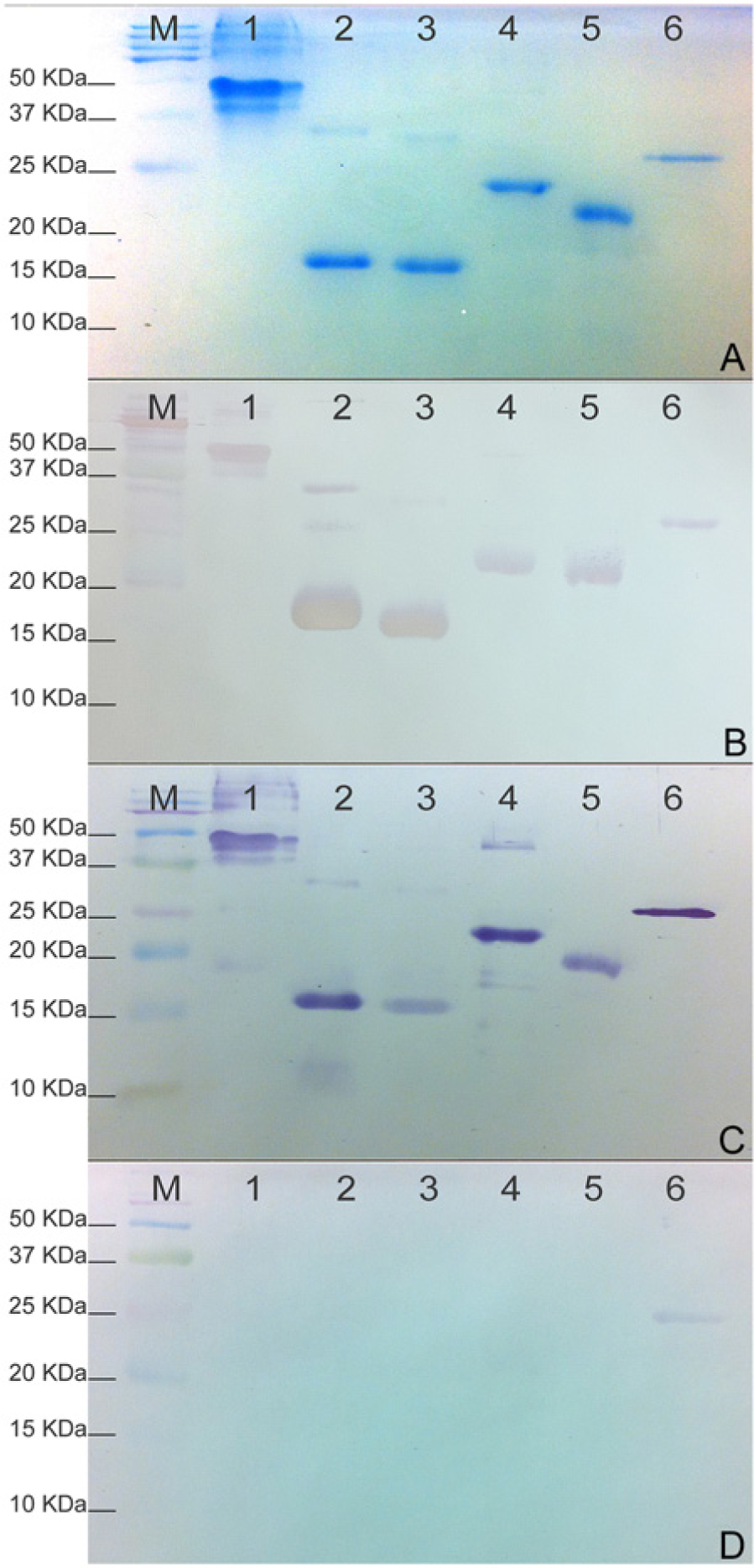
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot of the 6 purified, immunoreactive proteins.
Optimization of MIA
A total of 40 µg of GAPN (protein fragment of MSU_0029 [GAPNf]), 125 µg of MSU_0184 (pf184), and 5 µg of GrpE (MSU_0474) antigens were determined to be the optimal protein concentrations for bead coupling (bead ID 29, 62, and 86, respectively). A 1:300 dilution of the sera controls provided the highest dynamic range (7.03, 5.3, and 14.8, respectively) with the lowest negative background (411, 183.75, and 62.5, respectively). Optimal secondary antibody dilution was set as 1:200. Regardless of different protein concentrations (5, 25, 50, and 125 µg), MSU_0794, MSU_0110, and MSU_0108 protein fragment antigens showed consistently low dynamic range (<4) and high negative background (>1,300), which did not allow for the discrimination between low-positive and negative serum samples. Therefore, only antigens GAPNf, pf184, and GrpE were used to develop a three-plex assay for the detection of M. suis antibodies.
Cutoff limits for a negative test result were determined as being the average of the MFI plus 3 SD of sera from 20 M. suis–free, CDCD pigs. Accordingly, the average MFI for GAPNf was 361.30 (SD: 105.01), with an assay cutoff of 676.32; the average MFI for pf184 was 168.34 (SD: 64.54), with an assay cutoff of 361.97; and the average MFI for GrpE was 50.60 (SD: 37.87), with an assay cutoff of 164.23. Cross-reactivity between the coupled beads and the secondary antibody was negligible, as the background MFIs were consistently below 15 for all 3 antigens. The intra- and interassay variability of the assay as measured by the CV among replicates was also low (Table 2). No cross-reactivity between the antigens and sera from animals seropositive for various other pig diseases was observed, as MFIs were below the cutoff limits for each protein.
Intra- and interassay variability of the multiplex microbead immunoassay assay.*

“Negative” was a pool of 20 sera from cesarean-derived, colostrum-deprived pigs. “Acute” was a pool of serial sera from a splenectomized pig acutely infected with Mycoplasma suis. “Chronic” was a pool of sera from a non-splenectomized pig chronically infected with M. suis. Mean = mean of median fluorescent intensity. Numbers in parentheses are coefficients of variation.
Course of infection in the acutely and chronically affected pigs
The qPCR results for pigs 1 and 2 were reported elsewhere. 5 Along with the kinetics of the antibody response, the qPCR results are also referenced in Figure 2 for better comprehension. Interestingly, for pig 1, the MIA results were positive 17 days prior (day 18) to the detection of M. suis infection by qPCR (day 35; Fig. 2). The highest MFIs for all 3 antigens were observed at the first peak of bacteremia (day 46; GAPNf: 12,732.5; pf184: 4,437.5; GrpE: 6,326). Antigen pf184 showed borderline results on days 18 and 28 (day 18: 355.5; day 28: 350; cutoff for pf184 is 361.97) while the other 2 antigens were positive (day 18: 831.75; day 28: 1,048.5; cutoff for GAPNf is 676.32; day 18: 355.5; day 28: 443.5; cutoff for GrpE is 164.23). Except for antigen GrpE, subsequent bacteremic peaks did not induce an increased antibody response, and MFI continuously decreased. For antigen GrpE, MFI was 1,405.5 at the second peak of bacteremia, which increased to 2,523.25 at subsequent sampling and 2,817 at the last peak of bacteremia. Pig 2 had positive MFI results throughout the course of the infection; however, pf184 antigen decreased below the detection level by day 71 (day 71: 219.75; day 113: 294.5; day 121: 338; cutoff for pf184 is 361.97).
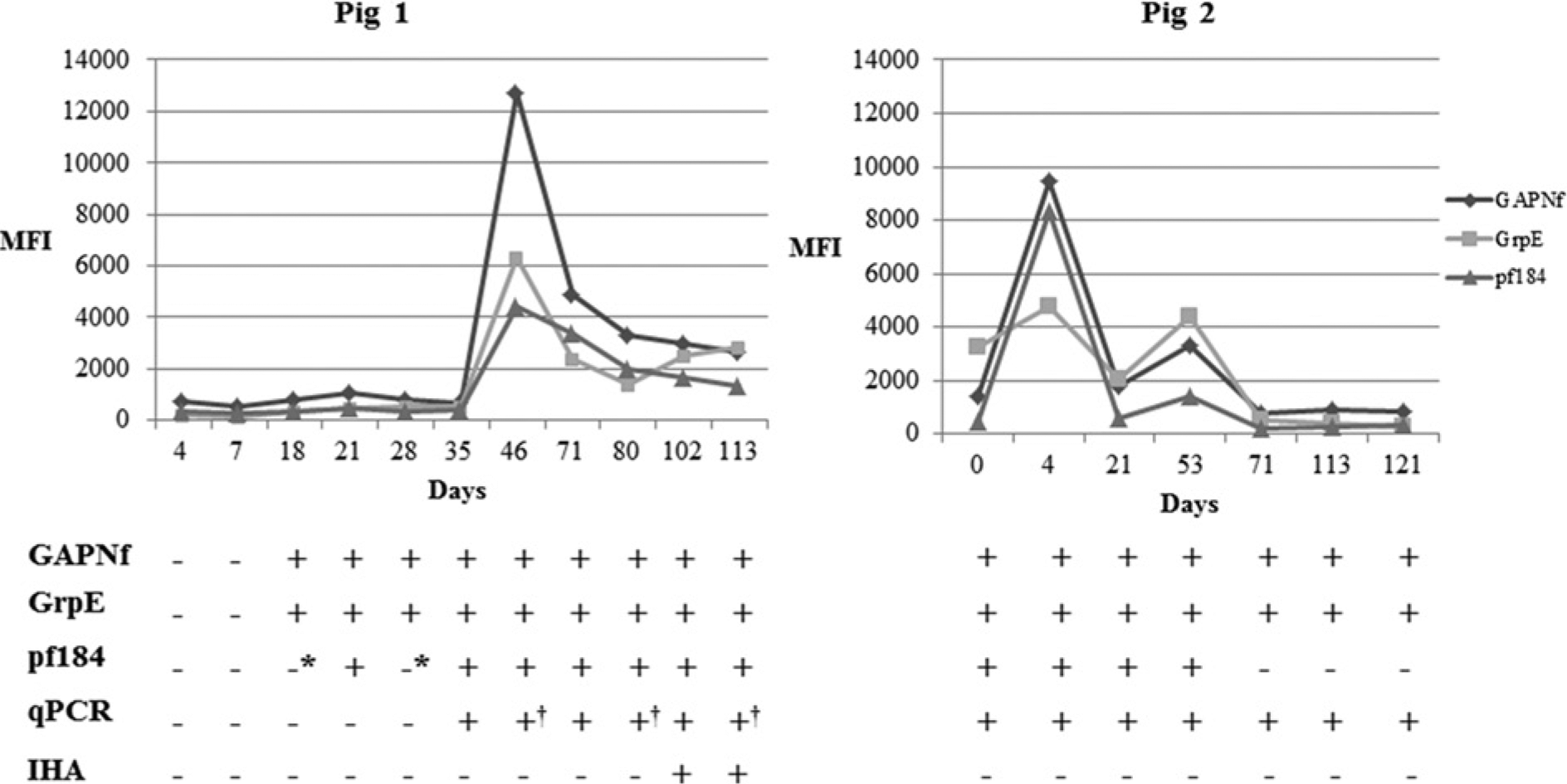
Kinetics of antibody response in pigs 1 and 2. Pig 1: splenectomized, acute disease model; pig 2: non-splenectomized, chronic disease model. MFI = median fluorescent intensity. *Borderline MFI results: 355.5 MFI in day 18 and 350 MFI in day 28 (pig 1). †Peaks of bacteremia in pig 1 (organisms seen on blood smears, bacterial load >1012 organisms/ml of blood, clinical signs were evident as described previously). 5
Field samples
Only 5 (9.8%) out of the 51 DNA samples were positive in the M. suis–specific qPCR, with bacterial load varying from 2.19 × 104 to 1.89 × 109 organisms/ml of blood. The MFI results for the serum samples are shown in Supplementary Figure S1. A total of 40 (78.43%) samples were positive for all 3 antigens, while only 4 (7.84%) samples were negative for all 3 antigens. Seven (13.72%) samples had discrepant serologic results: 4 were positive for 2 antigens (GAPNf and GrpE) and 3 were positive for only 1 antigen (GAPNf or GrpE; Supplementary Fig. S1); each of these samples was negative for antigen pf184. Of the 5 M. suis qPCR-positive samples, 4 were positive for all 3 antigens, while 1 was positive only for antigen GrpE (MFI: 309.5). Mean MFIs for qPCR-positive and -negative samples were not statistically different (p = 0.49 for GAPNf, p = 0.5 for GrpE, p = 0.81 for pf184).
Comparison between MFI and qPCR and IHA
Taken together (pig 1: n = 11; pig 2: n = 7; field samples: n = 51), a total of 69 sera were tested using IHA. Only 2 samples had positive results: sera from pig 1 at days 102 and 113 with titers of 1:160 and 1:80, respectively. The kappa coefficient between MFI and IHA was poor (κ = 0.01; no. of observed agreements: 12/69, 17.4%). Similar results were obtained for comparison of MFI and qPCR (κ = 0.07; no. of observed agreements: 26/69, 37.7%). However, when considering only pigs 1 and 2, agreement was considered to be good (κ = 0.684; no. of observed agreements: 16/18, 88.9%).
Discussion
The current study reports the identification of M. suis antigens and the development of a three-plex MIA that can be used for the detection of antibodies against M. suis. The overall success for identifying useful immunogenic targets for the assay was 23%. Thus, while 6 out of 13 putative antigens were immunogenic, only 3 of these were suitable serologic targets for the MIA. The diagnostic usefulness of the unsuitable targets in other immunoassays (e.g., ELISA) cannot be completely ruled out as certain antigens may behave differently depending on the serologic method employed.1,21 Interestingly, the 3 previously described M. suis immunogenic proteins (GAPDH, DnaK, and Ppa) were not detected in the library screening in the current study. The presence of UGA codons within these genes may have hampered their expression and subsequent detection in the study. In addition, it is possible that the different detection methods used in previous studies might account for the identification of different M. suis antigens.8,10
The antigens used in the MIA were the enzyme GAPN (GAPNf), the heat-shock protein GrpE, and a hypothetical protein (pf184) from a PGF. The GAPN protein is of particular interest because it is absent in other porcine mycoplasmas sequenced to date. It has been speculated that this enzyme might play a role in the survival of M. suis in the face of oxidants that are ubiquitous in its blood environment. 4 On the other hand, the GrpE is universally present in bacteria and has been described as an antigen in several pathogens but not in mycoplasmas.12,18,29 The serologic results for both GAPNf and GrpE antigens were similar with only 3 out of 69 samples having a positive result for only GAPNf or GrpE. Significant differences in the positive rate between 2 conserved proteins, GAPDH and DnaK, used in 2 different ELISAs have been reported. 7 The ability of the microbead approach to multiplex proteins and maintain equivalent stringent conditions for both GAPNf and GrpE might explain why such differences were not observed in the current study.
Although the antigen pf184 was initially considered suitable for the MIA, sample testing showed that this protein is a weak immunogen. MSU_0184 is part of the third largest PGF of M. suis strain Illinois, with 16 protein members. 4 Certain PGF have been associated with antigenic variation in other mycoplasmas, 2 and further study should be done to confirm if this mechanism also occurs in M. suis PGFs. It is possible that pigs 1 and 2 responded to a variant of the same PGF, generating only low-affinity antibodies against pf184. Thus, results for this antigen were negative both earlier and later in the course of infection. This antigen also detected fewer positive field samples when compared to GAPNf and GrpE. The possibility that full-length gene expression might increase the detection of positive animals cannot be discounted; however, the likelihood of antigenic variation makes this target less attractive for diagnostic purposes.
In contrast to previous studies,7,8 the antibody response, as measured by MFI, was the highest at the first peak of bacteremia for pig 1 (acute disease model). A previous study has reported that ELISA optical density values decline significantly and below cutoff limits during peaks of bacteremia in domestic pigs. 7 In the same study, 7 it was speculated that during acute clinical disease there is a derailment of immune response against the bacterium. Since the antibody response of only 1 acutely infected animal was analyzed in the current study, the possibility that differences in host immune responses might account for these discrepant findings cannot be discounted. Another possibility is that M. suis strain KI3806 20 may be more virulent than M. suis strain Illinois. Nonetheless, for both pigs in the present study, high MFIs appear to be associated with recent infection. Pig 2 was admitted into the experimental facilities when she was only 45 days old. It is possible that this pig had recently been infected with M. suis at the farm, showing a peak MFI on day 4, which declined thereafter. The evaluation of additional animals is needed to further clarify whether high MFIs might be used to identify a recently infected animal.
The high numbers of field pigs in the current study with antibodies to at least 2 M. suis antigens (44/51, 86.3%) was somewhat surprising given that most of these pigs were negative by qPCR. In study published in 2007, 2 immunodominant proteins (GAPDH and DnaK antigens) were used individually in ELISAs to detect antibodies against M. suis in sera of slaughterhouse pigs. 7 A high seropositivity rate (80.0%) was only observed in qPCR-positive samples, while among qPCR-negative samples, only 26.7% were serologically positive. 7 One possible explanation for the difference between studies is that the pigs in the current study were exposed to M. suis but have cleared the infection, whereas the pigs in Germany 7 failed to clear the organism. Alternatively, bacterial blood loads could have been below the detection limit of the qPCR in the present study. This seems an unlikely possibility given that a few as 2 hemoplasma organisms/µl of blood are detected in this assay. 5 In the future, understanding whether or not these animals eliminate infection and, if they do, how this is accomplished may help in the design of preventive strategies, such as vaccination.
False negatives by IHA have been previously described in both experimentally and naturally infected pigs. 6 In addition, ELISAs using purified crude antigen or recombinant Ppa were found to have greater numbers of positives when compared to IHA.11,15 In the current study, the only IHA-positive samples were from pig 1, late in the course of infection (102 and 113 days). The MFI values at these times, while still positive, had waned significantly from peak levels (day 46). Based on these findings and previous results in the literature,11,13,15 the IHA appears to be an insensitive method for detecting exposure to M. suis in pig herds. There is a need for the development of MIA, qPCR assays, and ELISAs in reference laboratories worldwide to facilitate the diagnosis and to better understand the extent of M. suis exposure and infection in pigs.
In the current study, serum samples from various experimentally or sham-infected animals were not available to evaluate the MIA sensitivity and specificity. However, the analytical specificity was tested using sera from animals that were seropositive for 5 common pig pathogens: M. hyopneumoniae, PCV-2, PPV, PRRSV, and PRCV. The MFI for each of the antigens was below the negative cutoffs established in the assay in the present study, confirming the absence of serologic cross-reactivity to these pathogens. Furthermore, the inclusion of the protein GAPNf, which is not present in other porcine mycoplasmas sequenced to date, contributes to the assay specificity. Supporting the high sensitivity of the assay are the great number of seropositive field samples and consistent antibody detection in the course of infection in pigs 1 and 2. Taken together, the findings of the current study suggest that the MIA is sensitive and specific for detection of antibodies against M. suis. By multiplexing these antigens, each may serve as a confirmatory test of the other, likely improving its overall efficiency.
In conclusion, knowledge of the antigenic core of M. suis has been expanded by the discovery of 6 novel antigens in the current study. Highly conserved bacterial proteins as well as certain hypothetical proteins from PGF have been shown to elicit host immune responses. The immunogenic PGFs identified in the current study can now be analyzed regarding antigenic variation as well as their role in evading the host immune responses and establishing chronic infection. The developed MIA for M. suis permits independent recognition of several specific antigen–antibody reactions and is a reliable tool for serologic diagnosis of M. suis exposure. Owing to the high capacity of MIA for multiplexing, other antigens of M. suis or those from various other pig pathogens could be added in the future, providing for a fast and simultaneous infectious disease detection panel.
Footnotes
Acknowledgements
The authors are grateful to Dr. Roman Pogranichniy for the CDCD pig samples, and Drs. Naíla C. do Nascimento and Yuefeng Chu for carefully reviewing this article.
a.
Protein A HP spin trap, GE Healthcare Technologies, Piscataway, NJ.
b.
QIAamp DNA mini kit, Qiagen Inc., Valencia, CA.
c.
Blood & cell culture DNA midi kit, Genomic-tip 100/G, Qiagen Inc., Valencia, CA.
d.
New England Biolabs Inc., Ipswich, MA.
e.
Lambda ZapII predigested vector kit with Gigapack Gold packing extract, Stratagene Corp., La Jolla, CA.
f.
NZ amine broth, Sigma-Aldrich, St. Louis, MO.
g.
3,3′,5,5′-tetramethylbenzidine (TMB) liquid substrate system for membranes, Sigma-Aldrich, St. Louis, MO.
h.
Phase lock gel, Eppendorf North America, Hauppauge, NY.
i.
Rockland Inc., Gilbertsville, PA.
n.
Champion pET100 directional TOPO expression kit with BL21 Star (DE3) E. coli, Invitrogen Corp., Grand Island, NY.
o.
QIAprep spin miniprep kit, Qiagen Inc., Valencia, CA.
p.
HisPur cobalt purification columns, Thermo Fisher Scientific, Waltham, MA.
q.
Rabbit anti-pig IgG, HRP-conjugated, Rockland Immunochemicals, Gilbertsville, PA.
r.
MicroPlex microspheres, Luminex Corp., Austin, TX.
s.
Rabbit biotin conjugated anti-pig IgG, Rockland Immunochemicals, Gilbertsville, PA.
t.
Phycoerythrin conjugated streptavidin, Rockland Immunochemicals, Gilbertsville, PA.
u.
Luminex 100, Luminex Corp., Austin, TX.
v.
xPONENT software, Luminex Corp., Austin, TX.
w.
GraphPad Software Inc., La Jolla, CA.
x.
Precision plus protein kaleidoscope standards, Bio-Rad Laboratories, Hercules, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Support was provided from Hatch Act Formula Grant, project no. IND020395. Funding support for PhD studies of A. M. S. Guimaraes was provided by the Brazilian Ministry of Education through Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fulbright Commission, Fulbright-Capes Scholarship Program (CAPES-Fulbright Program ID 167307/6). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.