
Abstract
The purpose of this study was to characterize light and electron microscopic findings from 9 dogs that had consumed aflatoxin-contaminated commercial dog food from recalled batches. Four dogs died and 5 were euthanized after signs of liver failure. Analysis of feed and liver samples confirmed exposure to aflatoxin. Of the 9 dogs, 8 had classic signs of liver failure, and 1 had signs of liver failure. Enlarged, pale yellow livers were seen macroscopically at necropsy in the dogs with subacute hepatopathy, and cirrhosis was noted in the dog with chronic hepatopathy. Histopathologic findings included hepatic lipidosis, portal fibroplasia, and biliary hyperplasia, which supported a diagnosis of subacute toxic hepatopathy in the 8 symptomatic animals. Marked lobular atrophy, bridging portal fibrosis, and regenerative hepatocellular nodules characterized the dog with chronic hepatopathy. Electron microscopy revealed marked hepatocellular lipid vacuolation and early fibroplasia in the dogs with acute hepatopathy and marked fibrosis and regeneration in the dog with chronic hepatopathy. Analysis of feed for aflatoxin consistently revealed high levels of aflatoxin B1 (range of 223–579 ppb), and hepatic tissue contained elevated levels of aflatoxin B1metabolite M1 (0.6–4.4 ppb). Although dogs are not commonly affected by aflatoxicosis, they are highly susceptible and can present with classic signs of acute or chronic hepatopathy. Characteristic gross, histologic, and electron microscopic changes help pathologists determine a presumptive toxic insult. Detecting aflatoxins or their metabolites in feed or liver specimens can help confirm the diagnosis of aflatoxicosis.
Introduction
Aflatoxicosis has been documented in humans, cats, dogs, rodents, pet birds, cattle, and poultry after exposure to feed contaminated with a mycotoxin-producing fungus. 4,6,10,12 15 Aflatoxin outbreaks are more common in livestock than in dogs. The last large canine outbreak in the United States was in 1998 6 The current case series resulted from an outbreak associated with mycotoxin-contaminated corn processed into dry commercial dog food. 18 This outbreak affected animals throughout the eastern United States in late 2005 through early 2006. Although a previous publication included some of the same dogs, 18 the purpose of this report is to describe the gross, histologic, and electron microscopic findings from the dogs in more detail, thereby extending the published findings.
Materials and methods
Animals
Nine dogs from the local area (including the cities of Benton, Athens, and Knoxville, TN) were presented to the University of Tennessee College of Veterinary Medicine (UTCVM) over the course of an aflatoxin outbreak that lasted several months. For several weeks before presentation, all the dogs had eaten the same brand of dog food, a some batches of which had been recalled because of recognized aflatoxin contamination. Batch and lot numbers from empty or partially empty feed bags were examined and determined to match the recall information on the company's Web site, confirming potential contamination. Historical information and physical examination findings are summarized in Table 1. All animals presented for treatment through the Small Animal Clinical Sciences Department. Significant clinical laboratory data (complete blood cell count, clinical chemistry, and coagulation profiles) are summarized in Table 2. Subsequent to unsuccessful therapy, animals were presented to the Pathology Service for necropsy and routine histopathologic examination.
Historical data on the 9 dogs affected by aflatoxin.
F = female; M = male; N = neutered; S = spayed.
Although most of the aflatoxin-exposed dogs presented as a cluster of cases in a 6-week time span, dog 9 presented to UTCVM approximately 8 weeks later (Table 1). Another mixed-breed dog from the same household had died at an emergency clinic 10 weeks earlier because of acute hepatopathy. Dog 9 died at home 8.5 weeks after initial presentation to the referring veterinarian and was submitted for a necropsy examination at UTCVM.
Pathology
At necropsy, all visceral organs and brain tissues were collected in 10% buffered formalin, processed for routine histopathology, and stained with hematoxylin and eosin. Masson trichrome and Pearls iron stains were used to confirm fibrosis or iron deposition, respectively. Five dogs (1, 2, 3, 7, and 9) had liver samples fixed in dual-purpose fixative (Carson). b These samples were processed for routine electron microscopy. PCR for leptospirosis was performed on hepatic samples from dogs 1 and 2. 16
Toxicology
Feed samples from feed bags from which 8 of the dogs were fed were submitted to another laboratory c for determination of aflatoxin (B1 and B2) levels by the ELISA method 19 or thin-layer chromatography (TLC), followed by high-performance liquid chromatography (HPLC). For mycotoxin detection, dog food samples were ground in a blender before being weighed and extracted for mycotoxins. Milli-Q H2O was added, followed by 100 ml of ethyl acetate d and acetonitrile d before filtration and aliquoting. Final samples were injected on a calibrated gel permeation chromatography column e and dried. Samples that tested positive for aflatoxins by TLC were selected for confirmation and quantification. The detection limit of HPLC quantitation was 0.004 ppm.
Frozen liver samples from all dogs were sent to another laboratory f for the determination of aflatoxin concentrations. Initially, canine liver samples were extracted and screened by TLC for aflatoxins B1, B2, G1, G2, and M1. 17 Specimens found to contain aflatoxin M1 by the initial TLC screen were further processed by TLC to purify the M1 for analysis by HPLC. All solvents used were reagent or HPLC grade, g and aflatoxin standards were purchased. h One-gram equivalents were spotted on a Silica Gel 60 TLC plate, i along with 0.3 ng and 0.5 ng aflatoxin M1 standards. The TLC plate was developed using chloroform:acetone:2-propanol (85:10:5, v:v:v). After development of the TLC plate, the plate was dried and viewed under long-wave ultraviolet light. Each area correlating to aflatoxin M1 was circled and scraped off individually into 2-dram vials, in addition to a blank area for background. Samples were eluted off TLC scrapings by adding 1 ml of methanol:water (90:10, v:v) to each vial for at least 2 hours. The solvent was removed to a clean vial and labeled appropriately. The methanol/water extracts were evaporated under nitrogen and derivatized for HPLC. Dried samples were derivatized by adding 100 μl trifluoroacetic acid (TFA) h to each vial and vortexing for 30 seconds. The TFA was evaporated under nitrogen, and samples were redissolved into 100 μl methanol. A 10-μl portion of each sample was injected using a 2695 Waters Separation Module j equipped with a Phenomenex Bondclone 10 C18 column (300 × 3.9 mm) k connected to a 2475 Waters Fluorescence Detector j using Ex 365 Em 440 gain 100. The mobile phase was water:-methanol:2-propanol (320:136:16, v:v:v:) at a 1 ml/min flow rate with a retention time for aflatoxin M1 at 5 min. Samples and standards were quantified against a standard curve of TFA-derivatized aflatoxin M1 for 0.3 ng, 0.5 ng, and 1 ng. Samples were corrected for loss from elution off the TLC plate. The limit of detection was 0.3 ppb, and the limit of quantitation was 0.5 ppb.
Clinical pathology data from 9 affected dogs. *
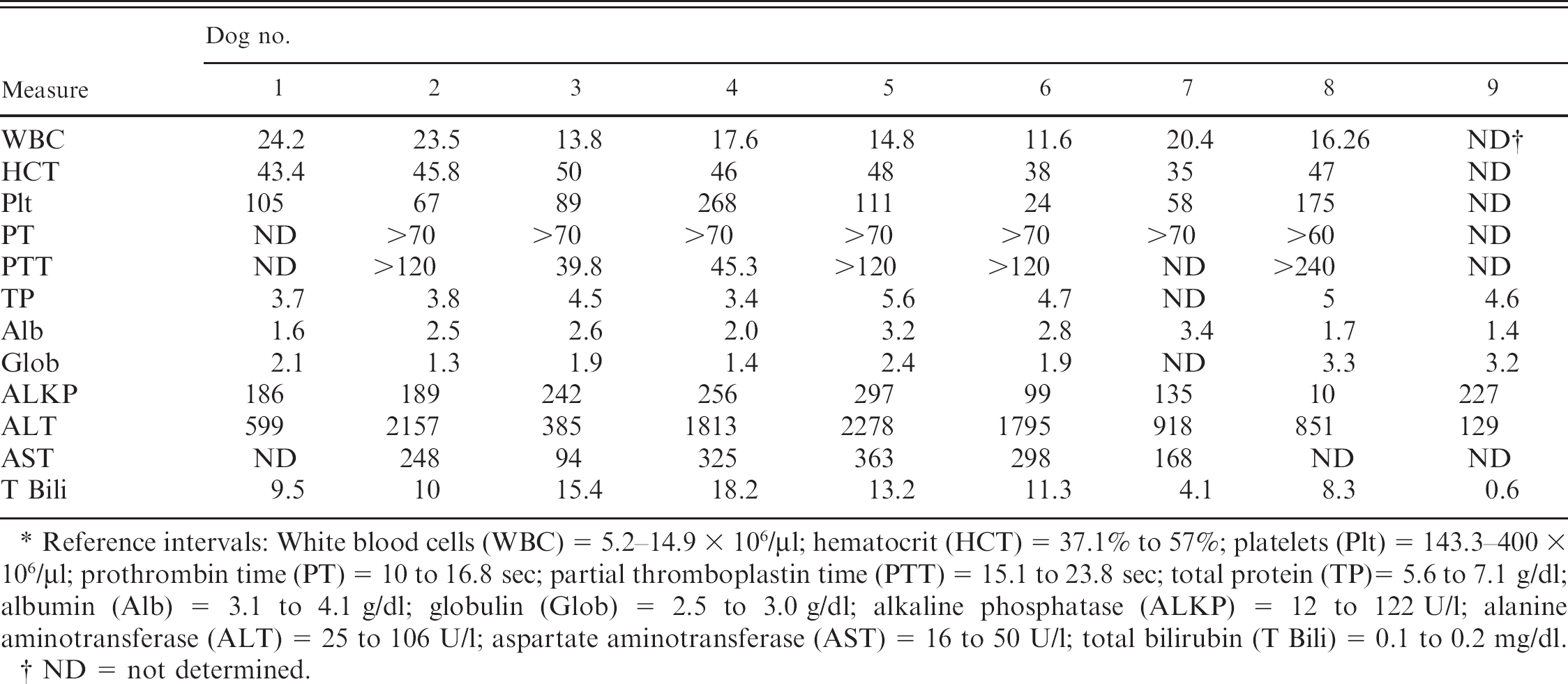
Reference intervals: White blood cells (WBC) = 5.2-14.9 × 106/μl; hematocrit (HCT) = 37.1% to 57%; platelets (Plt) = 143.3-100 × 106/μl; prothrombin time (PT) = 10 to 16.8 sec; partial thromboplastin time (PTT) = 15.1 to 23.8 sec; total protein (TP)= 5.6 to 7.1 g/dl; albumin (Alb) = 3.1 to 4.1 g/dl; globulin (Glob) = 2.5 to 3.0 g/dl; alkaline phosphatase (ALKP) = 12 to 122 U/l; alanine aminotransferase (ALT) = 25 to 106 U/l; aspartate aminotransferase (AST) = 16 to 50 U/l; total bilirubin (T Bili) = 0.1 to 0.2 mg/dl.
ND = not determined.
Most common gross findings in 9 dogs with aflatoxicosis. *

+ presence of lesion.
Results
Necropsy findings are summarized in Table 3. All dogs with hepatic failure had enlarged, pale yellow friable livers (Fig. 1) and variable systemic icterus, consistent with aflatoxicosis. Dog 9 had an atrophic green liver that contained multiple, variably sized, regenerative nodules throughout all lobes.
PCR for leptospirosis was negative for dogs 1 and 2 (data not shown). Table 4 summarizes the light microscopic hepatic changes. For the most part, significant hepatic pathology consistent with either subacute hepatopathy or chronic hepatopathy (dog 9) was evident. Subacute changes were characterized by varying degrees of hepatic cytoplasmic vacuolation throughout all 3 zones of the acinus (Fig. 2). Binucleate or multinucleate periportal hepatocytes were noted focally as clusters. Scattered individual hepatocyte necrosis and mild infiltrates of neutrophils and macrophages were associated with hepatocyte degeneration (Fig. 3). Additionally, the biliary epithelium was hyperplastic and prominent within and surrounding portal triads (Fig. 4). Reactive fibroblasts expanded and partially replaced the portal and periportal regions with variable amounts of fibrous connective tissue (Fig. 5), and there were small infiltrates of lymphocytes and fewer plasma cells. Midzonal hepatocytes contained moderate to large amounts of yellow/brown granular cytoplasmic pigment (bile). Larger bile ducts contained bile admixed with mucous plugs, and there was scattered sinusoidal extramedullary hematopoiesis.
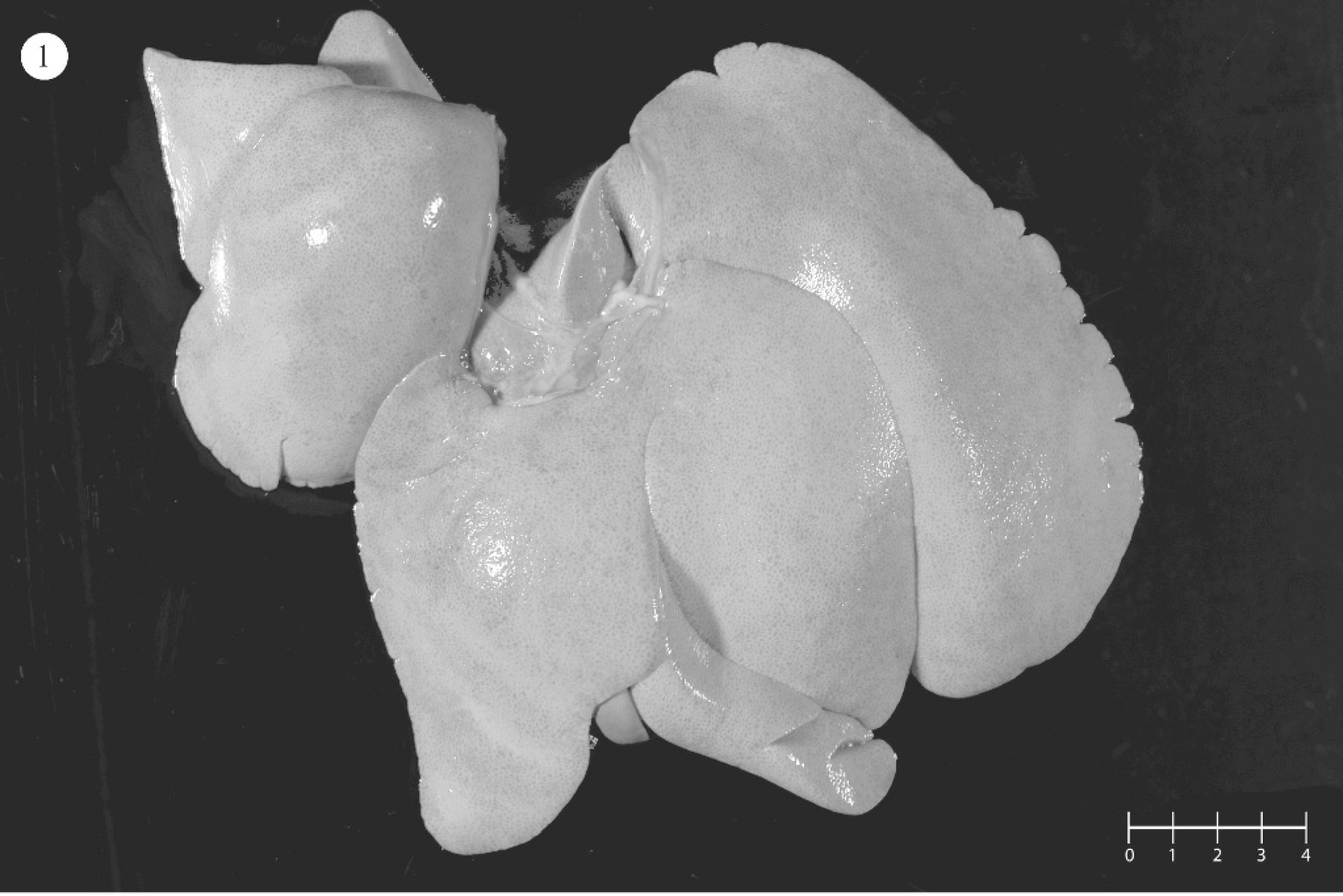
Gross photograph of an enlarged, friable, pale yellow liver consistent with subacute aflatoxicosis.
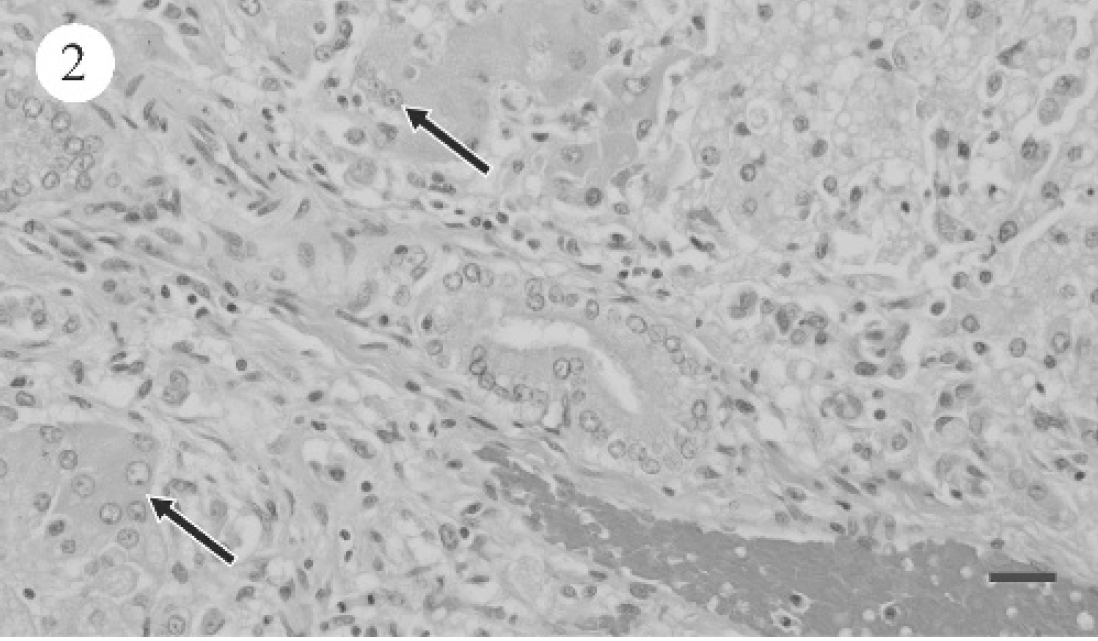
Photomicrograph of canine liver.
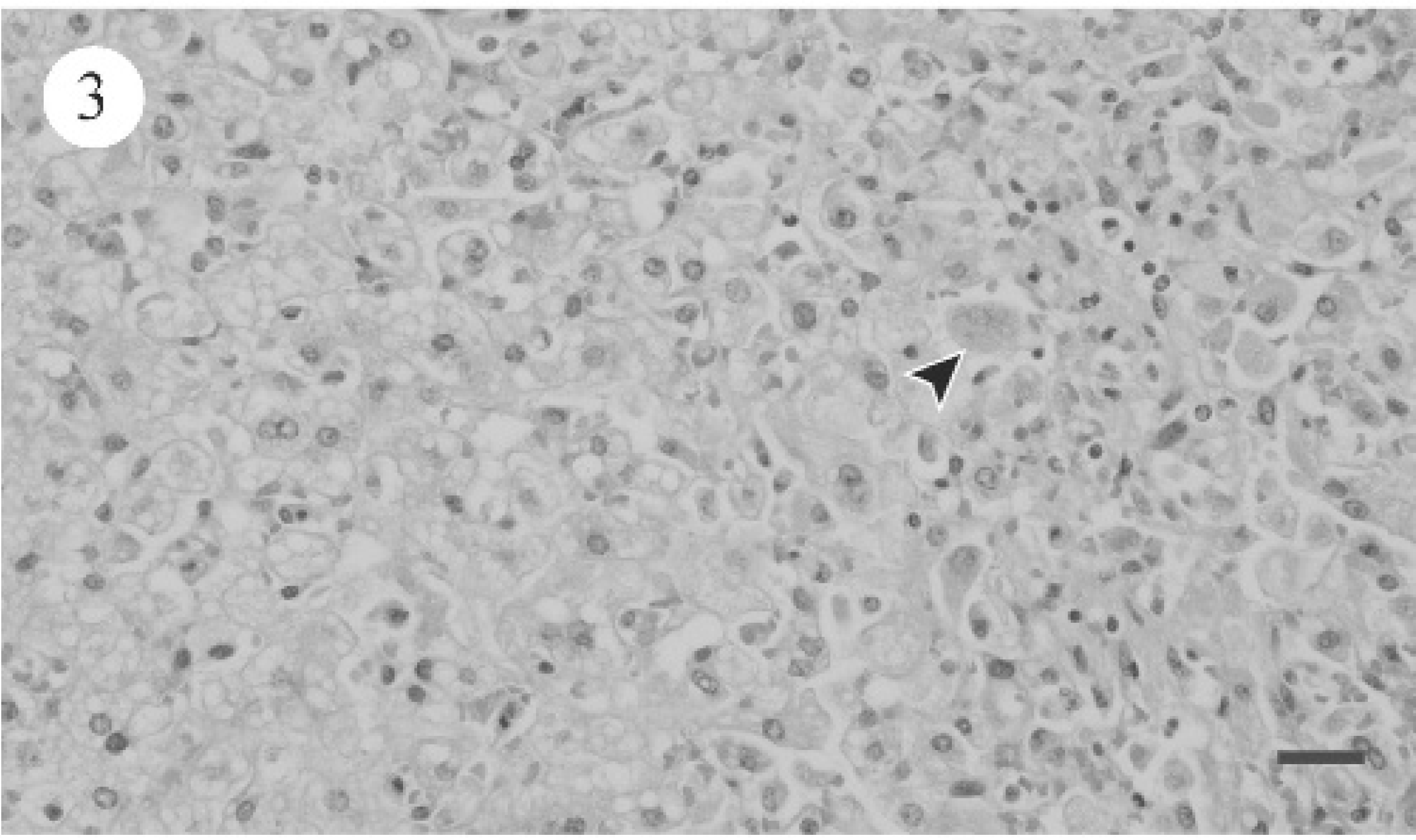
Photomicrograph of canine liver, showing diffuse, severe hepatocellular hepatic lipidosis and scattered hepatocyte necrosis (arrowhead). HE. Bar = 25 μm.
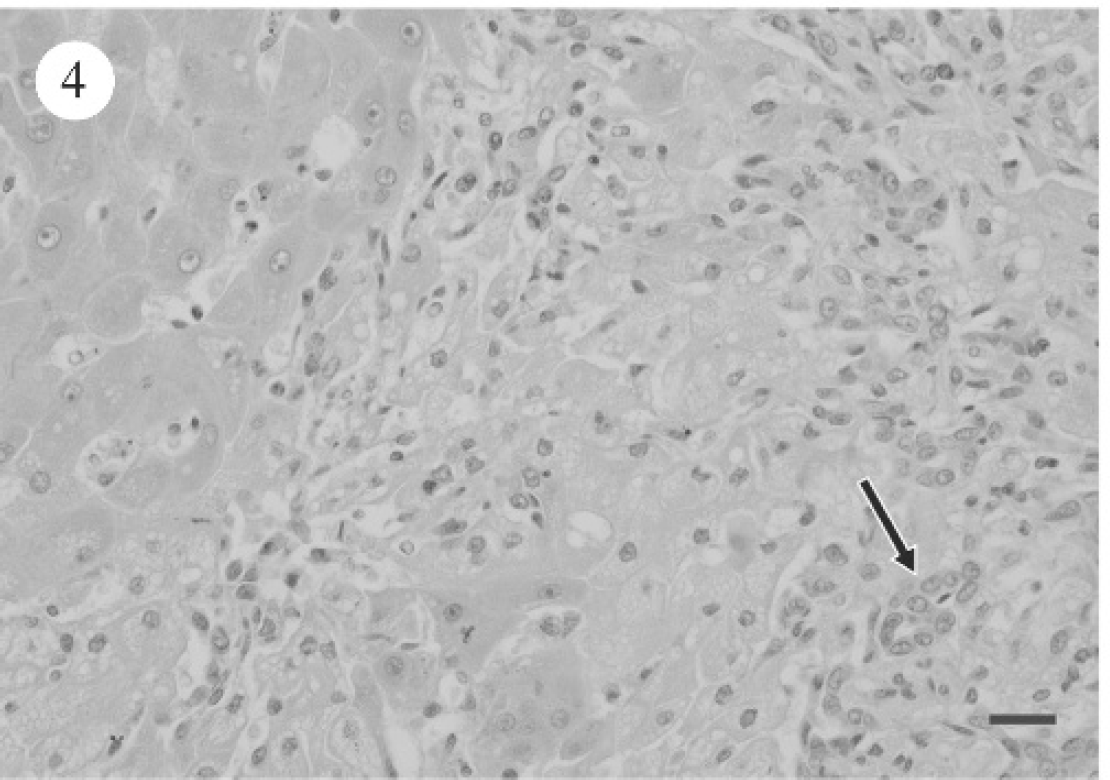
Photomicrograph of canine liver, showing moderate biliary hyperplasia (arrow) and portal fibroplasia. HE. Bar = 25 μm.
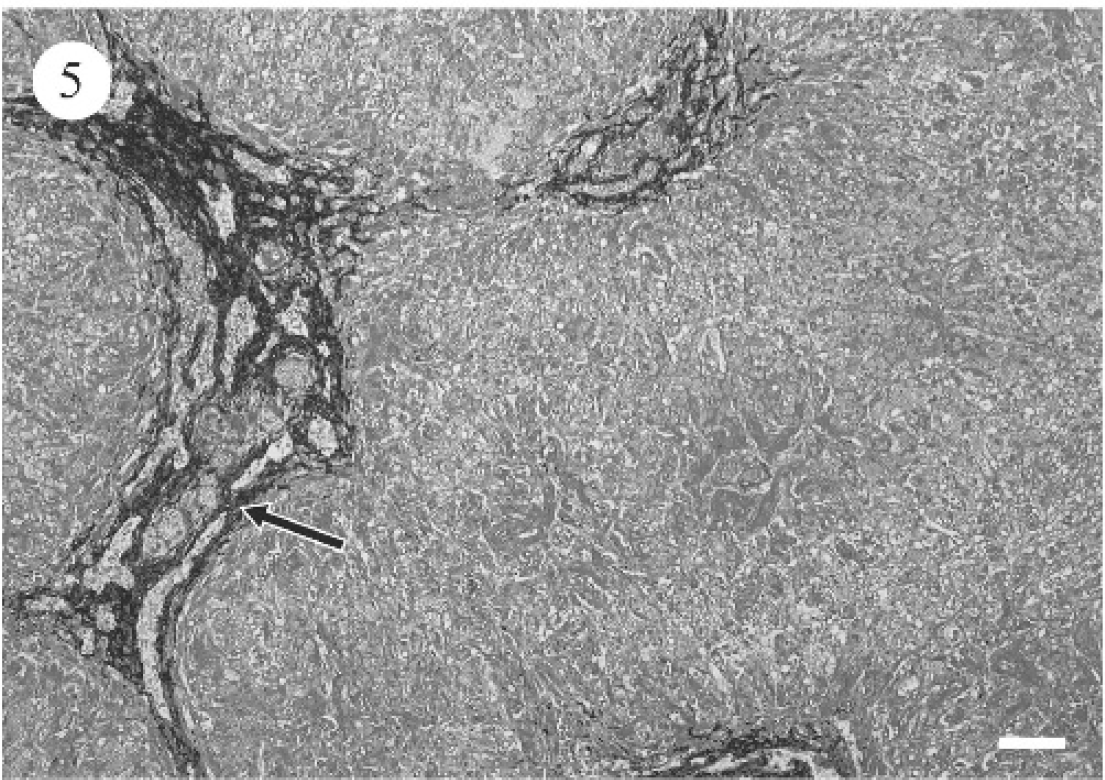
Photomicrograph of canine liver, revealing increased fibrous connective tissue within and extending from portal regions. Masson trichrome stain. Bar = 60 μm.
Most common hepatic histopathologic findings in 9 dogs with aflatoxicosis. *

+ = presence of lesion; EMH = extramedullary hematopoiesis; SMH = smooth muscle hypertrophy
Marked diffuse bridging portal fibrosis with infrequent retention of individual hepatocytes, moderate to marked bile ductule proliferation, and multifocal regenerative hepatocyte nodules characterized the chronic form. Large numbers of macrophages that contained golden green, granular, cytoplasmic pigment (presumptive bile, Pearls iron stain was negative for iron) and small numbers of lymphocytes and plasma cells were noted in the fibrotic regions. A mild, multifocal random coagulative necrosis of hepatocytes was present.
Significant electron microscopy lesions are summarized in Table 5. The primary changes noted in dogs with subacute hepatopathy were marked hepatocyte swelling and accumulation of intracytoplasmic lipids (Fig. 6A). Although collagen deposition was present, it was much less marked than in the chronic hepatopathy (dog 9) depicted in Figure 6B.
Levels of aflatoxin found in the associated feed samples are listed in Table 6. The tolerated concentration of aflatoxin in feed for dogs is less than 20 ppb (20 μg/kg). 3,5 Additionally, the liver aflatoxin M1levels determined by HPLC are listed in Table 7. Of the 9 dogs, 7 had detectable levels of metabolized aflatoxin in the liver. Aflatoxins B1, B2, G1, and G2were not detectable.
Discussion
The major toxin associated with aflatoxicosis is aflatoxin B1, the major effect of which is hepatotoxicity. 11 Lesser contributors are aflatoxin G1, aflatoxin B2, and aflatoxin G2. 7,11 Precise toxicity of moldy feedstuffs is impossible to assess without measuring toxin content in feed. 11 Food from feed bags from 8 of the dogs was tested for aflatoxin concentrations by HPLC (7 dogs) or the ELISA method (1 dog), the latter of which was performed on feed from the household of dog 8. Aflatoxin levels in feed were determined to be high for all 6 basset hounds (dogs 2 through 7) from the same household, the Australian shepherd (dog 1), and the Airedale terrier (dog 8). The concentrations of aflatoxin in most feed samples from this case series were consistently higher (598 ppb) than those noted in 2 similar diet-based epidemics in dogs where the food contained aflatoxin levels ranging from 100 to 300 ppb. 4,6 Although it is difficult to determine the exact length of time the dogs were fed contaminated feed, it was a relatively short period (weeks) compared with the required 90 to 120 days noted for manifestation of clinical signs of liver disease in a previous outbreak in Texas. 6 The varying rates of metabolism between different species, ages, nutritional status, and hormone levels hinder assessment of exposure in animals. 7 Additionally, the susceptibility of individual dogs can be affected by levels of sex hormones, age, dose, and/or degree of feed refusal. 18 Although the Food and Drug Administration suggests a zero tolerance for aflatoxin in food, it lists a legal limit of 20 μg/kg (ppb) in feed. 5,7 For dogs, the lethal dose, 50% (LD50) value is just 500 to 1,000 μg/kg (ppb), 9 and <60 μg/kg (ppb) is a toxic dose. 1,3,9,10
When tissue levels of aflatoxin were determined for all animals, the toxin was detected in 7 dogs, all of which had clinical, laboratory, and necropsy findings that supported a diagnosis of hepatic failure and histologic findings that suggested subacute toxic hepatopathy consistent with aflatoxicosis. In the dog with chronic hepatopathy (dog 9), the liver levels of metabolized aflatoxin were undetectable (<0.5 ppb), likely as a result of the 7-week period in which the contaminated feed was no longer being fed and existing aflatoxin metabolites were likely cleared. The low level of detectable aflatoxin in 1 of the 6 symptomatic basset hounds (dog 8) was harder to explain, especially as the features of the case were identical to those of the other basset hounds. Aflatoxin M1 may well have been cleared from the liver in the 7 days since the dog had stopped being fed the contaminated dog food. Although the half-life of aflatoxin in dogs has not been determined, in turkeys where aflatoxin B1 feeding at 500 ppb was discontinued, the half-life of aflatoxin was 1.4 days. 8 This suggests that even a delay of several days in testing for aflatoxins after cessation of feeding the affected food could alter aflatoxin metabolite levels in tissue.
Electron microscopy findings from 5 dogs with aflatoxicosis.
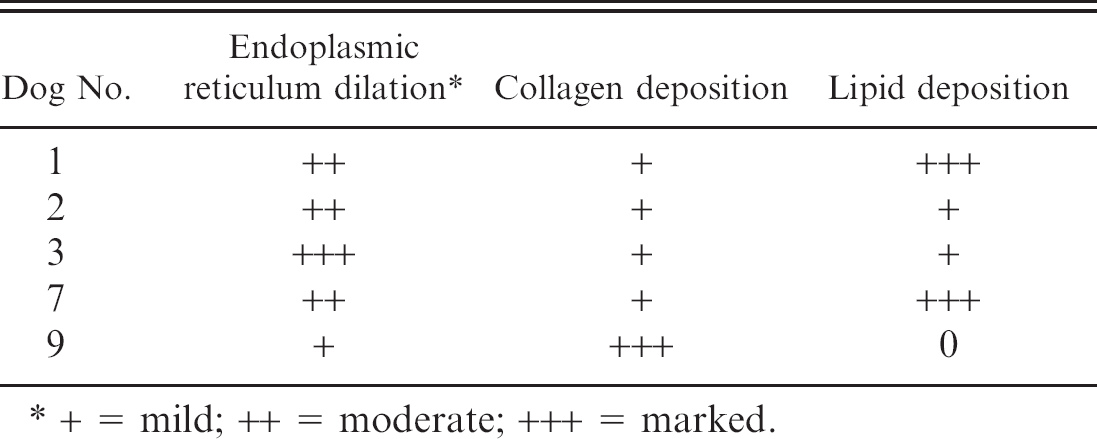
+ = mild; ++ = moderate; +++ = marked.
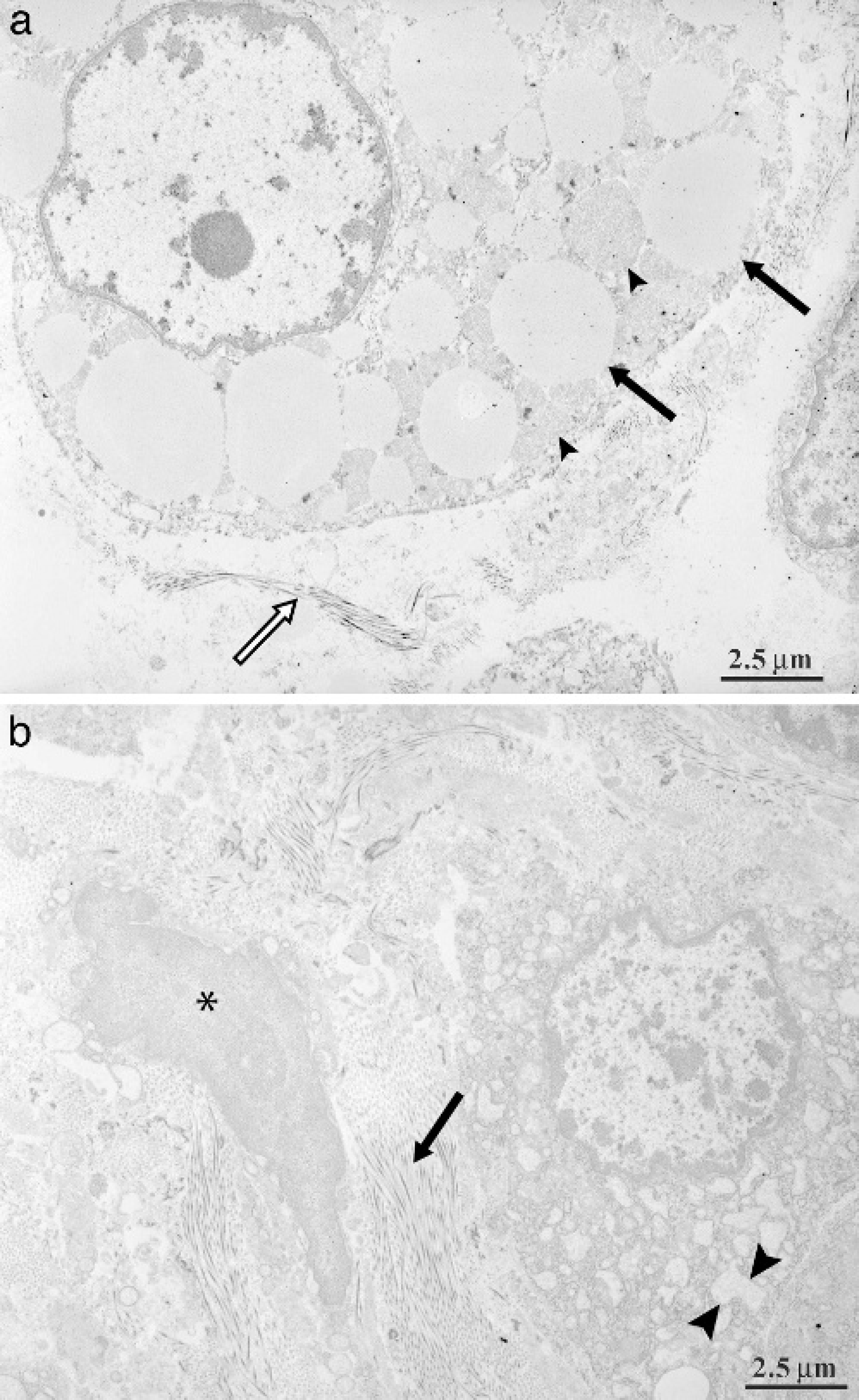
Transmission electron micrograph of liver from a dog with subacute aflatoxicosis. A, marked cytoplasmic accumulation of fat vacuoles is noted (arrows). Numerous mitochondria are also present (arrowheads). A small amount of collagen deposition is noted extracellularly (white arrow). Bar = 1.0 μm.
Results of aflatoxin analysis of dog food by HPLC. *
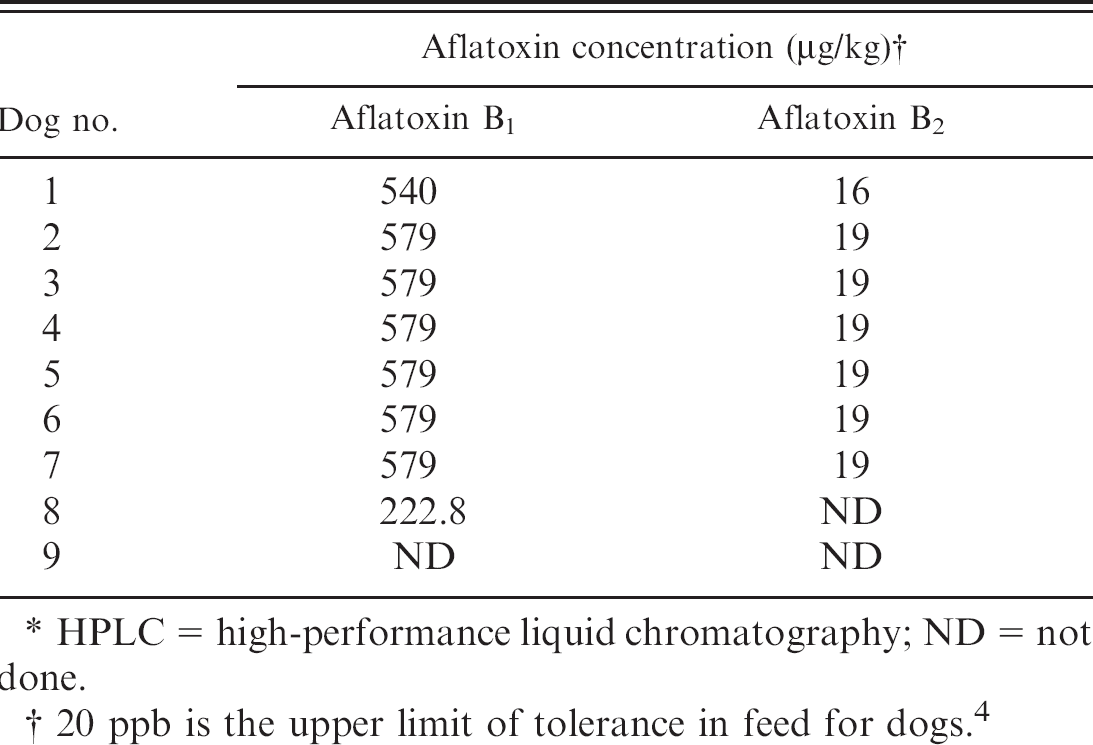
HPLC = high-performance liquid chromatography; ND = not done.
20 ppb is the upper limit of tolerance in feed for dogs. 4
All the dogs with confirmed high levels of aflatoxin in the feed samples had clinical, laboratory, and necropsy findings that supported a diagnosis of hepatic failure and histologic findings and electron microscopy findings that suggested subacute toxic hepatopathy. 4,11 The differential list of toxins in this case series was limited because of the known food exposure, but other toxins, such as pyrrolizidine alkaloids, microcystins, acetaminophen, ibuprofen, and infectious agents such as Leptospira could be considered. The presence of aflatoxin M1 in fresh or frozen liver samples collected from suspected cases of aflatoxicosis indicates aflatoxin exposure.
The liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST), as well as total bilirubin, were greatly elevated in the vast majority of the dogs (See Table 2). The degree of ongoing hepatocyte necrosis and swelling would predispose the dogs to elevated ALT and AST levels after leakage from the cytosol. Additionally, hepatocellular swelling, cholestasis, and portal fibrosis with biliary hyperplasia would predispose the dogs to hyperbilirubinemia. These changes were not noted in the dog with chronic hepatopathy.
Aflatoxin M1 levels derived from high-performance liquid chromatography analysis from frozen liver samples of all 9 dogs.
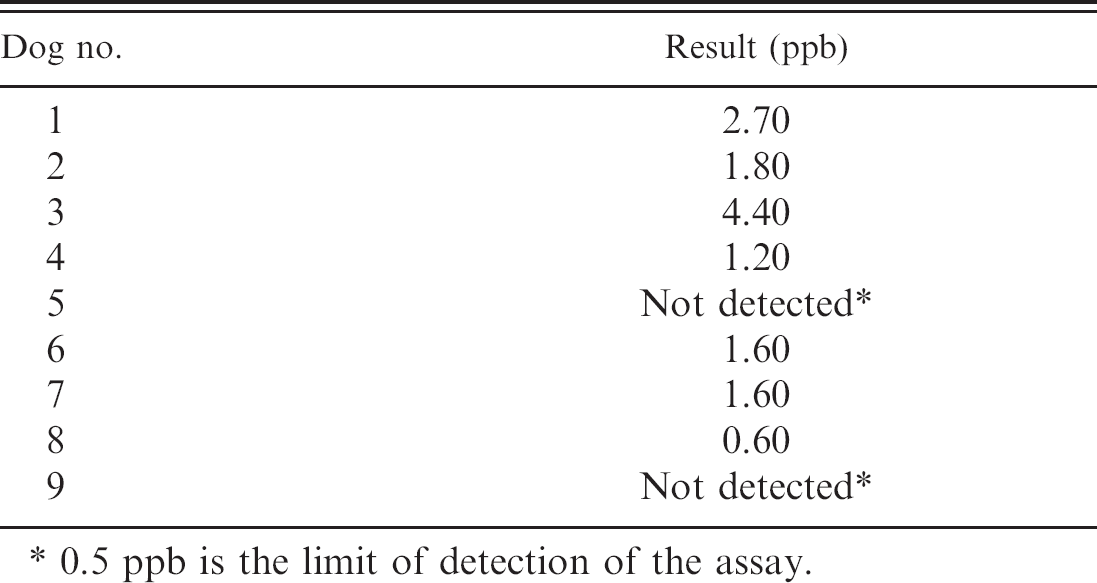
0.5 ppb is the limit of detection of the assay.
After aflatoxin exposure, affected dogs often present with liver failure and occasionally hemorrhagic diatheses. 3,6,7,10,14,15 A large percentage of the dogs in this report had hemorrhagic diatheses characterized by thrombocytopenia, prolonged activated partial thromboplastin times, and/or one-stage prothrombin times. In one report, hemorrhagic diatheses occurred in 60% of aflatoxin cases. 4 Theories for the clotting abnormalities include the fact that aflatoxins are coumarin-like and act as anticoagulants. 7 Hemorrhagic tendencies can occur as sequelae to overall decreased liver function and deficient hepatic production of prothrombin or fibrinogen. 4,7 The stimulus for disseminated intravascular coagulation in liver disease is a combination of release of thromboplastic substances from damaged hepatocytes 4 and potentially decreased clearance of these substances because of underlying liver failure. 7
Unlike hemorrhagic diatheses, the association of hepatic vascular shunting, characterized by central-vein, smooth-muscle hypertrophy, and aflatoxicosis has not been previously documented. It is hard to know whether the presence of venous arterialization makes the dogs more susceptible to aflatoxin exposure or whether it is an acquired change related to the presence of altered blood flow through livers affected by fibrosis. Because of the rapid clinical course in most animals in this study, it would seem that the smooth muscle hypertrophy was preexistent and not a response to toxicity. Three dogs had lesions of arterialization of the central veins; 1 dog with these lesions had chronic aflatoxicosis (dog 9). Occurrence of multiple vascular channels around central veins and interlobular vessels and perivascular accumulations of lymphocytes and Kupffer cells have been previously noted in dogs with aflatoxin exposure. 2 Veno-occlusive disease is part of the aflatoxicosis syndrome in cattle, 2,4 but its significance is unknown, and it has apparently not been reported in dogs.
The livers from affected dogs were enlarged as a result of hypertrophy of the hepatocellular smooth endoplasmic reticulum and discolored pale yellow because of bile retention and some degree of fatty vacuolar change within the cytoplasm. 6,7,11 Aflatoxin accumulation causes depletion of glycogen within hepatocytes by 24 hours and increased lipid vacuoles by 72 hours. 13 Additionally, in more chronic cases, lesions include increased firmness because of fibrosis, fine nodular regenerative hyperplasia, edema of the gallbladder wall, and bile-tinged ascites. 10,11,14
The range of nonhepatic gross lesions that have been associated with previous outbreaks of canine aflatoxicosis include hydroperitoneum, hydropericardium, hydrothorax, pulmonary congestion, pulmonary edema, lymph node edema, anasarca, subcutaneous edema, and less commonly, glomerulopathy. 9 Other changes manifested as part of aflatoxin-induced hepatic failure include widespread petechiation, gastric ulcers, melena, intestinal hemorrhage, hematomas, icterus, reactive bone marrow, lymph node atrophy, and splenomegaly due to red pulp hyperplasia. 4 In a previous study, ascites was seen in 6 of 10 cases and was thought to be a sequelae to portal fibrosis and vascular hypertension or to hypoproteinemia secondary to decreased protein production by a diseased liver. 4 Of the 9 dogs, 7 had hypoproteinemia. Additionally, all 9 dogs had marked icterus. However, in one previous study, icterus was identified in only 20% of affected animals, possibly as a result of the duration of aflatoxin exposure or stage of the disease process. 4 Only 1 of the 9 dogs in this study had a marginally reduced hematocrit, whereas up to 70% of cases in another study were reported to be anemic. 4
The histologic lesions observed in the dogs reported herein are similar to those that have been previously described for aflatoxicosis. 4,11 Bastianello et al. 4 described the pathology of acute, subacute, and chronic aflatoxicosis. The subacute form is typically characterized by extensive bile ductule hyperplasia intermingled with varying degrees of fibrosis. 4 Additionally, there is normal to slightly disrupted architecture with indistinct lobulation, extensive fatty degeneration in all regenerating hepatocytes, and bridging fibrosis. 4,10 12 Based on this description, all but 1 of the 9 dogs had subacute hepatopathy; 1 had the chronic form (dog 9). Typically, the chronic form is characterized by extensive fibroplasia. Additionally, there is severe bile ductule proliferation and visible regenerative hepatocellular nodules. 4
Electron microscopy done on 6 dogs showed changes similar to those described previously in pigs with aflatoxicosis. 13 To the author's knowledge, electron microscopic findings in dogs with aflatoxicosis have not been published previously. Previous porcine changes included cell disruption, mitochondrial swelling, lipid droplet accumulation, and extensive dilation of the endoplasmic reticulum within the centrilobular and midzonal hepatocytes. 13 Lipid droplet accumulation, dilation of endoplasmic reticulum within hepatocytes, and collagen deposition were the most prominent changes in the canine cases described here (Table 4).
The last dog (dog 9) to be affected by the outbreak died 7 weeks after feeding of the contaminated diet ceased. This is a considerably longer survival than the 3-week containment period determined in a previous outbreak in Texas. 6 Unfortunately, food from this household was not tested, but bull mastiffs from the same home were clinically afflicted. With intensive therapy, all these dogs survived, showing either variance in susceptibility between dogs within the same home or a variance in exposure.
Although widespread canine aflatoxicosis cases are rare, the cases presented here represent a geographic cluster (including the cities of Athens, Benton and Knoxville, TN) of cases of aflatoxicosis in dogs exposed to contaminated commercial dog food. The clinical, laboratory, and necropsy data supported hepatic failure. Microscopic and electron microscopy findings supported subacute toxic hepatopathy. Feed analysis of samples obtained from feed bags with matching batch and lot numbers from the manufacturer recall supported aflatoxin exposure. Detectable aflatoxin metabolite levels from livers also provided support for aflatoxicosis in these animals. In cases of hepatic failure with bleeding tendencies where infectious etiologies can be eliminated, acute aflatoxicosis should be considered, especially if there is a known point source or a geographic cluster of cases with similar dietary intake.
Acknowledgments
The following pathologists oversaw necropsy examinations on several of the dogs: Dr. Rebecca Moore, Dr. Danielle Reel, Dr. Stephen Yeomans, Dr. Robert Donnell, and Dr. Alison Tucker. Ms. Misty Bailey is acknowledged for editorial assistance, Mr. Shane Cummings for gross specimen photography, and Ms. Anik Vasington for graphic assistance.
Footnotes
a.
Diamond Pet Food, Gaston, SC.
b.
StatLab Medical Products, Lewisville, TX.
c.
Diagnostic Center for Population and Animal Health, College of Veterinary Medicine, Michigan State University, East Lansing.
d.
Burdick and Jackson, Muskegon, MI.
e.
OI Analytical, Columbia, MO.
f.
Veterinary Diagnostic Laboratory, College of Veterinary Medicine, Iowa State University, Ames.
g.
Fisher Scientific, Fairlawn, NJ.
h.
Sigma, St. Louis, MO.
i.
EM Separation Technology, Gibbstown, NJ.
j.
Waters Corp., Milford, MA.
k.
Phenomenex, Torrance, CA.