
Abstract
A polymerase chain reaction (PCR) assay was developed for the detection of Mycoplasma dispar in nasal mucus samples collected from calves. The target DNA sequence was the 16S rRNA gene, and the fragment was selected within a region of high polymorphism. The specificity and detection limit of the method were determined. This method was then used for the detection of M. dispar in nasal swabs collected from 301 calves, including 155 clinical samples from animals showing signs of respiratory disease and 146 samples from healthy animals. PCR with generic primers was applied to the detection of Mollicutes, followed by the detection of M. dispar. Mollicutes were detected in 52.05% of clinical samples from healthy animals and in 90.96% of samples from sick animals. Mycoplasma dispar was detected in 6.16% of healthy animals and in 34.84% of sick animals. The PCR assay was useful in verifying the presence of M. dispar in calves and may be a useful tool in monitoring this mycoplasma in cattle herds.
Keywords
Mycoplasma dispar is frequently isolated from animals with respiratory disease but may also be detected in healthy animals. 9 This species has been isolated in different countries and is usually transmitted from infected to healthy animals through respiratory secretions. 10 Mycoplasmas are fastidious, slow-growing microorganisms that require complex media for their growth. Thus, molecular techniques are necessary for their diagnosis.
The following microorganisms were used in the present study: Mycoplasma bovis Donetta, M. dispar M29/79, Mycoplasma mycoides subsp. mycoides SC PG1, M. mycoides subsp. mycoides LC Y-goat, Mycoplasma bovirhinis PG43, Mycoplasma bovoculi M165/69, Mycoplasma bovigenitalium PG11, Mycoplasma alkalescens PG51, Mycoplasma canadense 275C, Mycoplasma arginini G230, Mycoplasma verecundum GM893, Mycoplasma cynos H831, Mycoplasma edwardii PG24, Mycoplasma caprine SM12, Mycoplasma felis CO, Mycoplasma ovipneumoniae Y-98, Mycoplasma pulmonis PG34, Mycoplasma mycoides capri PG3, Mycoplasma equifoetale N93, Mycoplasma salivarium PG20, Mycoplasma fermentans PG18, Mycoplasma gallisepticum PG31, Mycoplasma synoviae WUU1853, Mycoplasma conjuntivae HRC581, Mycoplasma flocculare Ms42, Mycoplasma capricolum Calif kid, Mycoplasma collis 58B, Mycoplasma hyopneumoniae J, Acholeplasma laidlawii PG38, Ureaplasma diversum ATCC 449783, Ureaplasma urealyticum T960, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 10145, Staphylococcus aureus ATCC 25293, Streptococcus pyogenes ATCC 12345, Klebsiella pneumoniae ATCC 13883, Salmonella typhimurium C5 CIP, Bacteroides fragilis C45A AUS 00306, B. vulgatus C18E AUS 00293, and Actinomyces viscosus ATCC 910144. The strains were obtained from Universidade de São Paulo and Universidade Federal Fluminense, Brazil. The species were cultured on modified Friis (FF) medium 3 and Ureaplasma medium 8 containing 5% CMRL-1066 (with glutamine and without bicarbonate a ). Other walled bacterial strains were included as controls and were cultured in brain heart infusion medium. b
The 16S rRNA gene of M. dispar deposited in GenBank under the accession number AF412979 was used as the target DNA sequence. The sequence was analyzed with the Megalign program c to identify target regions with genetic differences among mycoplasma species. The region showing the highest polymorphism was identified. Different primers complementary to this region were obtained using the PrimerSelect program. c The selected set of primers for subsequent synthesis had the following sequences: MDF 5′-TTA AAG CTC CAC CAA AAA-3′ and MDR 5′-GTA TCT AAA GCG GAC TAA A-3′ and yielded an amplicon of 433 base pairs (bp).
Different hybridization temperatures (50-60°C), MgCl2 concentrations (1.0-2.5 mM), and deoxyribonucleotide (dNTP) concentrations were tested during the standardization of the polymerase chain reaction (PCR). These parameters were varied while maintaining the Taq polymerase and buffer concentrations and the number of cycles constant. The specificity of the method was evaluated by amplifying Mycoplasma spp. DNA of bovine, avian, swine, and human origin, as well as DNA from cell wall-containing bacteria. In each DNA amplification, M. dispar M29/79 and sterile ultrapure water were used as positive and negative controls, respectively. The detection limit of the PCR assay was determined using 10-fold dilutions of DNA extracted from M. dispar M29/79. A total of 20 mL of culture was used for extraction of DNA by the boiling method. 2 The DNA extracted was quantified in a spectrophotometer 1 at an absorbance of 260 nm. Following this procedure, decimal dilutions of the extracted DNA ranging from 1 μg to 1 atto DNA/μL were prepared in sterile, ultrapure water. After each dilution, the samples were boiled for 1 minute and promptly homogenized to prevent the formation of DNA lumps.
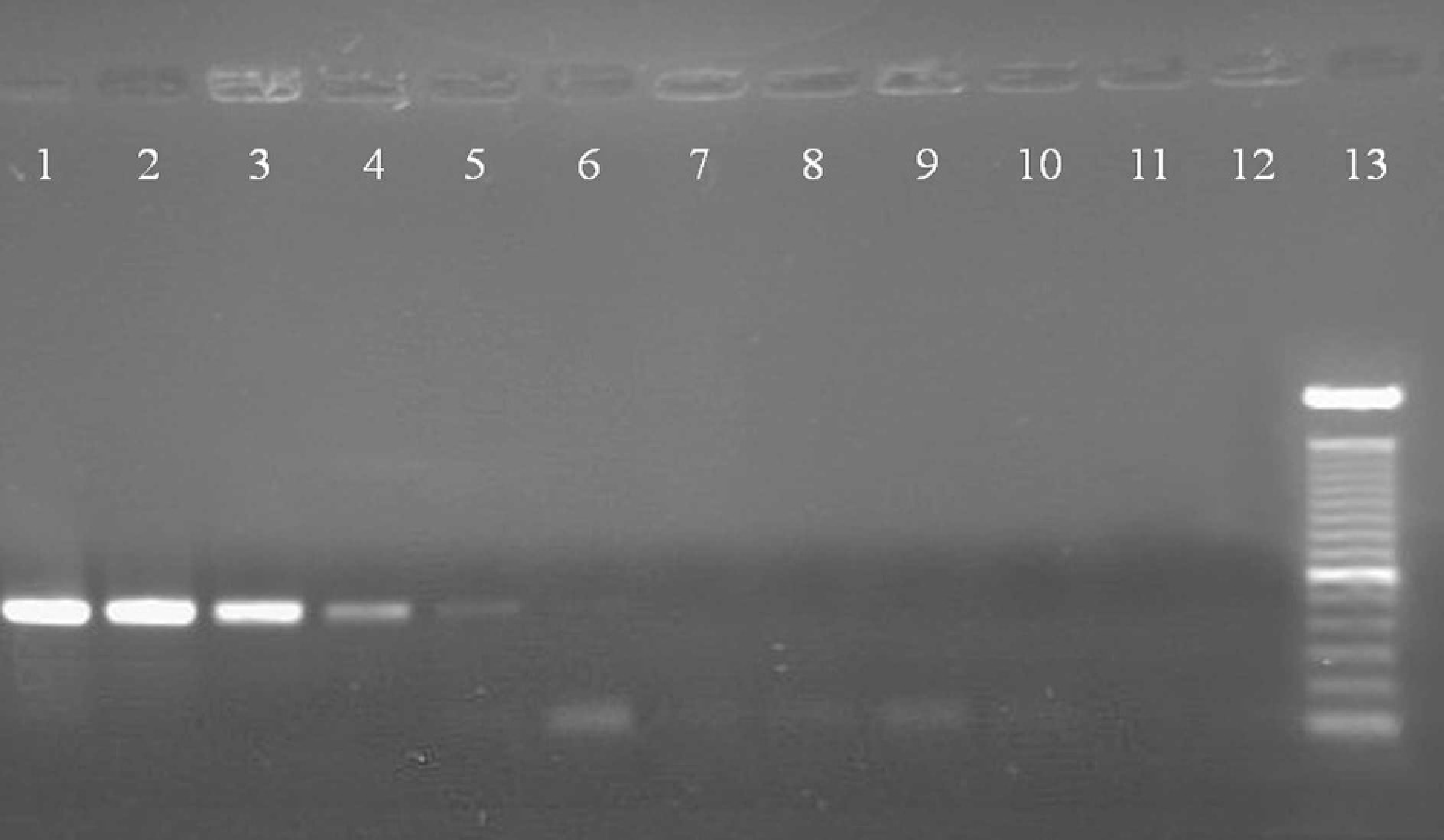
Detection limit of the PCR methodology for detection of M. dispar. Electrophoresis of PCR products from decimal dilutions of the extracted DNA from a culture of 20 mL of M. dispar. Lane 1:1 μg DNA/μL; Lane 2: 100 ng DNA/μL; Lane 3: 10 ng DNA/μL; Lane 4: 1 ng DNA/μL; Lane 5: 100 pg DNA/μL; Lane 6: 10 pg DNA/μL; Lane 7: 1 pg DNA/μL; Lane 8: 100 fg DNA/μL; Lane 9: 10 fg DNA/μL; Lane 10: 1 fg DNA/μL; Lane 11:1 atto DNA/μL; Lane 12: negative control (water); Lane 13: molecular size markers, 100 bp.
The reaction volume was 50 μL containing 5 μL PCR buffer (500 mM KCl, 200 mM Tris, pH 8.4), 50 mM of each dNTP (dATP, dCTP, dGTP, and dTTP), 2.0 mM MgCl2, 50 pmol of each primer, 2.0 μL of the chromosomal DNA to be tested, and 2 units Ampli Taq DNA polymerase. e The amplification reaction consisted of one cycle at 94°C for 5 minutes, followed by 35 cycles at 94°C for 1 minute, 53.6°C for 1 minute and 72°C for 1 minute, and a final cycle at 72°C for 5 minute. The amplified products were analyzed on a 1.5% agarose gel containing 0.5 μm/mL ethidium bromide in TAE buffer (40 mM Tris-acetate, 2 mM EDTA, pH 8.0). The products were visualized and photodocumented under ultraviolet light using a photodocumentation system. f
A total of 301 nasal swabs were collected from calves of both sexes, up to 1 year old (each sample representing 1 animal), from 10 different Brazilian farms (7 in the State of São Paulo, 2 in Minas Gerais, and 1 in Bahia). The samples were collected from animals showing signs of respiratory disease (n = 155) and from apparently healthy animals (n = 146). The animals with respiratory disease signs presented coughing, nasal discharge, fever, and pulmonary wheezing sounds. None of these signs was observed in apparently healthy animals. The samples were collected with a swab by rubbing the nasal cavity. The clinical material collected from each animal was stored and transported in 2 mL of liquid FF medium 3 at 4°C, and then incubated at 37°C for up to 4 days.
DNA was extracted from 1 ml of the clinical sample following the boiling method. Polymerase chain reaction was initially performed using the generic primers (GPO3 and MGSO) to detect Mollicutes as previously described. 10 Positive samples were retested using the M. dispar-specific primers. The fragments amplified by PCR using the MDF-MDR primers sequenced. Sequencing was performed in a MegaBACE 1000 apparatus. g The conditions for injection and electrophoresis were 2 Kv/60 sec and 6 Kv/230 min, respectively. The sequences obtained were compared with those deposited in the database of the National Center for Biotechnology and Information (NCBI) using the BLAST program. Species were identified based on the best alignment score.
The concentrations of 50 mM of each dNTP, 2.0 mM of MgCl2, and the annealing temperature of 53.6°C enabled the specific amplification of a product of approximately 433 bp of M. dispar. Nonspecific reactions were not observed under these conditions. The PCR threshold using the MDF/MDR primers was tested by preparing decimal dilutions of M. dispar DNA. As shown in Fig. 1, DNA of this species was detected at a concentration of 10 pg DNA/μL, corresponding to a dilution of 10−5 of 1 μg DNA/ml initial solution.
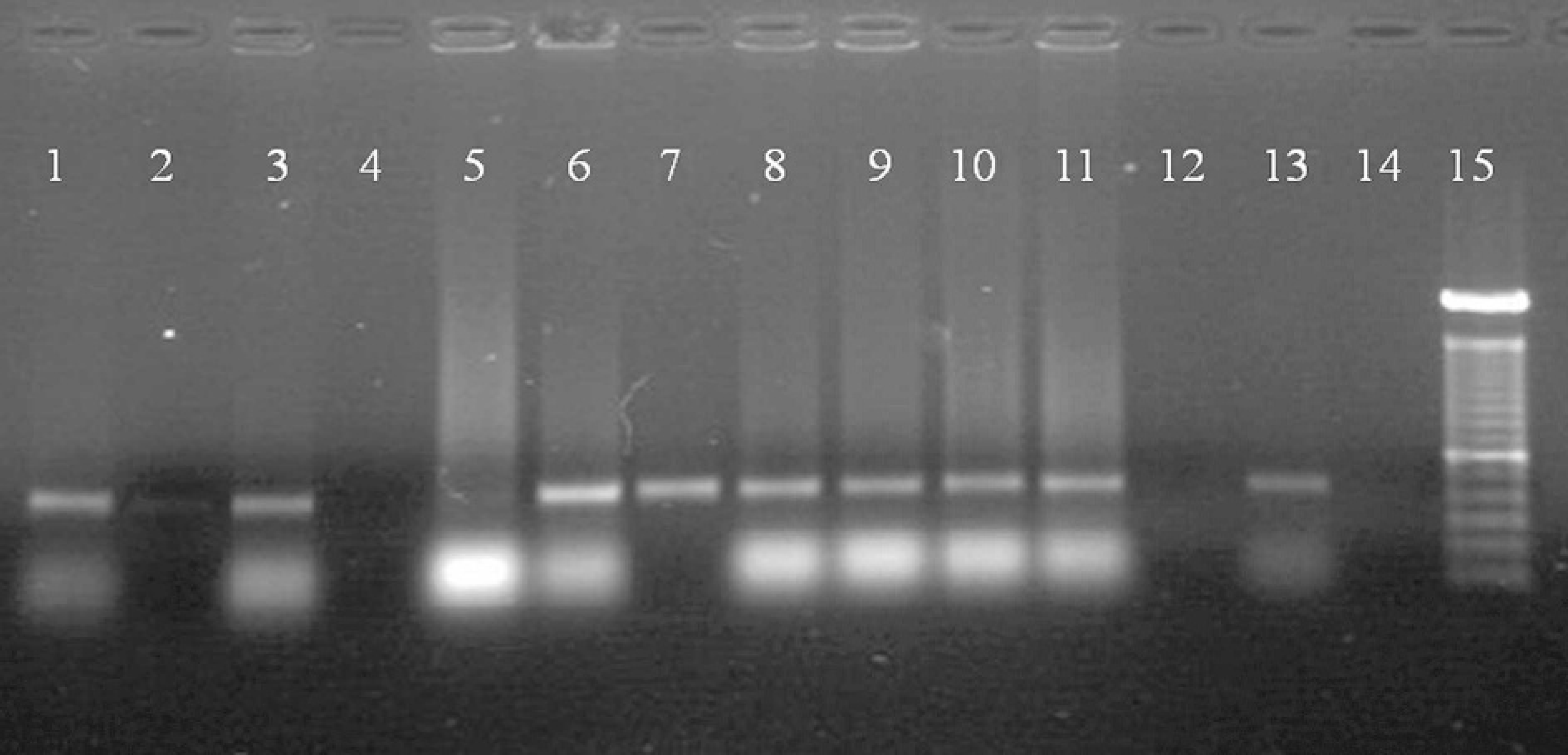
Electrophoresis of PCR products obtained with primers MDR and MDF to detect M. dispar in nasal samples of calves. Lanes 1 to 12: Nasal swab samples; lane 13: positive control (M. dispar); lane 14: negative control with water; lane 15: molecular size markers, 100 bp.
The PCR for Mollicutes detected these bacteria in 76 (52.05%) clinical samples from apparently healthy animals and in 141 (90.96%) samples from sick animals. M. dispar DNA was detected in 54 (34.84%) clinical samples from animals with respiratory disease and in 9 (6.16%) healthy animals (Fig. 2), with the frequency of detection of M. dispar being significantly higher in sick animals as compared to healthy ones (P < 0.05, chi-square test). All of the sequenced PCR products showed a sequence similarity of 98% to 99% with the M. dispar reference (GenBank Accession Number AF412979).
Culture is the reference procedure for the diagnosis of mycoplasmas; however, the slow growth of these bacteria makes this technique time-consuming even for specialized laboratories. In addition, M. dispar is one of the most difficult mycoplasma species to grow. Therefore, antibodies against M. dispar have been used as the only tool for the detection of this agent in some epidemiological studies. 7 Molecular diagnostic techniques enable rapid and specific detection of these microorganisms during the early stages of the disease in most types of mycoplasmosis. PCR is one of the most widely applied, sensitive, and efficient techniques in the detection of mycoplasmas. PCR assays for the detection of mycoplasmas generally target sequences on the 16S rRNA gene. 4,10 Some sequences are highly conserved and therefore useful for specific detection of mycoplasma species.
In the present study, the detection frequency of M. dispar by PCR was higher in cattle with signs of respiratory disease than in apparently healthy animals. Healthy animals that were positive for M. dispar by PCR had close contact with sick animals infected with this species. However, in farms where sick animals were absent, this microorganism was not detected. Similarly, other researchers have reported high frequencies of M. dispar isolation from postmortem samples. 1,5 Without isolation using specific media, using the same developed PCR showed that M. dispar is more frequent in sick calves compared to other bovine mycoplasmas (unpublished data, Timenetsky). Thus, the PCR test used in the present study efficiently detected M. dispar in live animals.
One other study has described a PCR for the detection of M. dispar; however, that study did not evaluate clinical samples. 6 The PCR assay reported herein was first used to detect M. dispar in pure culture, then tested against other Mycoplasma species, and finally, it was used with clinical samples. The successful use of the assay on clinical samples demonstrates its viability as an alternative diagnostic method in M. dispar control and surveillance studies.
Acknowledgements. This study was supported by FA-PESP (grant 03/02733-3). We thank Aricelma P. França for valuable technical assistance, and Prof. Dr. Elmiro Rosendo and Prof. Dr. Mario Julio Ávila-Campos for providing the reference strains.
Footnotes
a.
Gibco BRL-Life Technologies Inc., São Paulo, Brazil.
b.
Difco Laboratories, Detroit, MI.
c.
DNASTAR, Madison, WI.
d.
Beckman DU-600, Beckman Instruments Inc., Fullerton, CA.
e.
Invitrogen Life Technologies, São Paulo, Brazil.
f.
Vilber Lourmat, Marne-la-Vallée, France.
g.
Amersham Biosciences, Uppsala, Sweden.