
Abstract
Triple-negative breast cancers are the most aggressive subtypes with poor prognosis due to lack of targeted cancer therapy. Recently, we reported an association of A-kinase anchor protein 4 expression with various clinico-pathological parameters of breast cancer patients. In this context, we examined the effect of knockdown of A-kinase anchor protein 4 on cell cycle, apoptosis, cellular proliferation, colony formation, migration, and invasion in triple-negative breast cancer cells. We also examined the synergistic cytotoxic effect of paclitaxel on A-kinase anchor protein 4 downregulated triple-negative breast cancer cells. Knockdown of A-kinase anchor protein 4 resulted in significant reduction in cellular growth and migratory abilities. Interestingly, we also observed enhanced cell death in A-kinase anchor protein 4 downregulated cells treated with paclitaxel. Knockdown of A-kinase anchor protein 4 in cell cycle resulted in G0/G1 phase arrest. Knockdown of A-kinase anchor protein 4 also led to increased reactive oxygen species generation as a result of upregulation of NOXA and CHOP. In addition, levels of cyclins, cyclin-dependent kinases, anti-apoptotic molecules, and mesenchymal markers were reduced in A-kinase anchor protein 4 downregulated cells. Moreover, downregulation of A-kinase anchor protein 4 also caused tumor growth reduction in in vivo studies. These data together suggest that A-kinase anchor protein 4 downregulation inhibits various malignant properties and enhances the cytotoxic effect of paclitaxel, and this combinatorial approach could be useful for triple-negative breast cancer treatment.
Keywords
Introduction
Breast cancer is the second leading cause of cancer-related deaths in women worldwide. 1 Despite available treatment options for breast cancer, most of the treatment approaches are ineffective due to molecular heterogeneity and disease subtypes with poor prognosis. 2 Endocrine manipulation is the only therapy in hormone receptor–positive tumors. Tumors that are estrogen receptor (ER)/progesterone receptor (PR)-positive are much more likely to respond to hormone therapy than tumors that are ER/PR-negative. For patients with human epidermal growth factor receptor 2 (HER2)-positive breast cancers, targeted anti-HER2 antibodies have shown good prognosis. Triple-negative breast cancers (TNBCs) do not express ER or PR and lack HER2 overexpression and are the most aggressive subtypes that are difficult to treat. TNBC accounts for 15%–20% of all breast cancer with disproportionate number of metastatic cases and breast cancer deaths.3,4 The standard treatment for metastatic TNBC is the combination of paclitaxel and other cytotoxic drugs; however, to date, they have failed to demonstrate significant activity in metastatic breast cancer.5,6 Also, so far there is no targeted therapy for TNBC because of the absence of any targetable receptors.7,8
During the last decade, various chemotherapeutic agents alone or in combination have been reported to induce endoplasmic reticulum (ER) stress in tumor cells. 9 Persistent ER stress eventually initiates apoptosis, leading to cell death. 10 In this context, CHOP has been shown to act as ER stress effector that targets several downstream apoptotic genes, including NOXA. As a result of upregulation of apoptotic genes and NOXA, onset of apoptosis occurs, which leads to cell death. 11 Moreover, CHOP contributes to generation of reactive oxygen species (ROS) as well. Also, it has been documented that CHOP induces BIM, PUMA, and NOXA expression levels.12–14 NOXA, a pro-apoptotic member, belongs to the BCL-2 protein family, 15 plays a key role in apoptotic cell death,16,17 and is activated by various factors including ROS. Hence, CHOP/NOXA and ROS overproduction may be involved in the initiation of apoptosis and cell death.
Earlier studies on gene expression microarray analysis of various cancer cells revealed a restricted expression of novel gene, namely, A-kinase anchor protein 4 (AKAP4). 18 It has been also proposed that AKAP4 functions as a scaffolding protein and tethers cAMP-dependent protein kinase A (PKA) in various signaling modules and subcellular organelles. 19 Interestingly, PKA has been shown to be involved in the majority of human tumors and malignant properties that include cancer cell proliferation, angiogenesis, and chemoresistance.20–23 Recently, we reported an association of AKAP4 expression with various clinico-pathological parameters of breast cancer patients. 24 Our recent reports have put forth evidence that AKAP4 is associated with tumor progression in cervical, ovarian, and colon cancers.25–28 Recently, we demonstrated that knockdown of AKAP4 in ovarian cancer cells resulted in ROS generation, DNA damage, reduced cellular growth, proliferation, cell cycle arrest in vitro, and reduced tumor growth. 26 In this context, as monotherapy, recent clinical trials employing small interfering RNAs (siRNAs) against vascular endothelial growth factor (VEGF) and kinesin spindle protein (KSP) have shown promising results to treat metastatic endometrial and hepatocellular carcinoma. 29 Therefore, by employing gene silencing approach, we examined the involvement of AKAP4 in various molecular pathways contributing to TNBC growth. Attempts were also made to investigate the combinatorial effect of paclitaxel and knockdown of AKAP4 on various malignant properties, apoptosis, and epithelial–mesenchymal transition (EMT) pathways in TNBC cells. This current report opens up new avenues for bench-to-bedside translation and may contribute toward the development of novel treatment option for TNBC.
Materials and methods
Cell lines and culture
Breast cancer cell lines, triple-negative MDA-MB-231 (TNBC, highly metastatic basal, triple-negative ER− PR− HER2−), and MCF-7 (luminal A, ER+ PR+ HER2−) were used in this study. The cell lines were procured from American Type Culture Collection (ATCC, Manassas, VA) and were cultured in Dulbecco’s modified eagle media (DMEM) supplemented with 10% fetal bovine serum (FBS) according to recommended growth medium and culture conditions. All breast cancer cell lines were checked for mycoplasma contamination by mycoplasma polymerase chain reaction (PCR) detection kit (Applied Biological Materials, Inc., Richmond, Canada).
Western blotting and indirect immunofluorescence
AKAP4 protein expression was examined in breast cancer cells by indirect immunofluorescence (IIF) and Western blotting as described earlier.26,28 Micrographs were captured using the Carl Zeiss LSM 510 Meta confocal microscope (Zeiss, Oberkochen, Germany).
Antibodies
AKAP4 (ab56551); NOXA (MA141000); poly(ADP-ribose) polymerase (PARP) (46D); CHOP (MS1-250); β-actin (ab6276); Calnexin, ER marker (sc-70481); GM130, Golgi bodies marker (sc-55591); MTCO2, mitochondrial marker (ab3298); Lamin A/C, nuclear envelope marker (sc-7292); Pan-cadherin, plasma membrane marker (ab6528); BAD (sc-8044); BAX (B8554); BCL-2 (SAB2900350); Bcl-xL (B9429); BID (SAB5300512); caspase 3 (sc-56052); caspase 7 (sc-56063); caspase 8 (sc-70501); cytochrome-C (sc-13560); PUMA (ab9643); MCL-1 (ab32087); APAF1 (ab2000); cyclin A1 (sc-53233); cyclin B1 (sc-7393); cyclin D1 (sc-8396); cyclin E (sc-56310); p21 (sc-817); cyclin-dependent kinases (CDKs) (CDK1 (ab18), CDK2 (ab32147), CDK4 (sc-23896), and CDK6 (sc-7961)); proliferating cell nuclear antigen (PCNA; sc-25280); E-cadherin (ab1416); N-cadherin (ab76011); P-cadherin (ab19350); SLUG (ab51772), TWIST (ab50887); and Vimentin (ab92547). Secondary antibodies: anti-mouse IgG horseradish peroxidase (HRP; Jackson ImmunoResearch Laboratories, Baltimore, PA, USA, 115-035-003), anti-rabbit IgG HRP (Jackson ImmunoResearch Laboratories, 111-035-144), anti-mouse Texas Red (SAB3701111), and anti-mouse IgG fluorescein isothiocyanate (FITC; Jackson ImmunoResearch Laboratories, 115-095-003).
Knockdown of AKAP4 employing plasmid-driven gene silencing approach
Knockdown of AKAP4 was carried out employing plasmid-driven short hairpin RNA (shRNA) approach. AKAP4 shRNA constructs and scrambled negative control (NC) shRNA construct were procured from Sigma-Aldrich (St. Louis, MO, USA; Supplementary Table 1). Three AKAP4 shRNA constructs (shRNA1, shRNA2, and shRNA3) and the NC shRNA were used to ablate AKAP4 as described earlier. 26 Similarly, knockdown of NOXA and CHOP (target sequences; Supplementary Table 1) was done as described earlier. 28 Reverse transcription of total RNA was done as described earlier. 26 Quantitative PCR (qPCR) was performed as described earlier 28 using AKAP4 primers (Supplementary Table 2). Knockdown of NOXA and CHOP was also carried out employing NOXA shRNA and CHOP shRNA targets, which were gifted by Dr Subba Rao Gangi Setty, Department of Microbiology & Cell Biology, Indian Institute of Science, Bangalore, India.
Cellular proliferation, cell viability, and colony formation assay
To study the effect of knockdown of AKAP4 on various malignant properties of breast cancer cells, various assays were performed as described earlier. 26 Cellular proliferation was examined in cells transfected with NC shRNA or AKAP4 shRNA2 or shRNA3 as described earlier. 26 To examine cell viability, cells transfected with NC shRNA or AKAP4 shRNA2 or shRNA3 were seeded in a 96-well plate to carry out 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described earlier. 28 The experiments were carried out in triplicates three independent times.
Measurement of synergistic effect
Combinatorial effect of AKAP4 shRNA and paclitaxel in different combinations was examined in MDA-MB-231 cells as described earlier. 30 Combination index (CI) values were assessed using CompuSyn 1.01 software (ComboSyn, Inc., Paramus, NJ, USA). The experiments were carried out in triplicates three independent times.
Cell cycle analysis
Flow cytometric analysis was carried out to evaluate cell cycle arrest as described previously. 26 The data were obtained and analyzed by BD-FACSVerse (BD Biosciences, San Jose, CA, USA). The experiments were carried out in triplicates three independent times.
Scanning electron microscopy
To study the phenotypic changes, knockdown of AKAP4 was carried out in breast cancer cells as described earlier. 28 Scanning electron microscopy (SEM) images were captured by electron microscope (EVO LSM10 Zeiss, Germany) at 20 kV using Smart SEM software.
Annexin V staining
Onset of apoptosis as a result of AKAP4 downregulation with NC shRNA or AKAP4 shRNA2 or shRNA3 was examined using flow cytometric analysis after staining the cells with annexin V using Annexin-V-PerCP-Cy5-5-A staining kit (BD Biosciences) as described earlier. 28
TUNEL assay
As a result of AKAP4 downregulation, DNA damage was examined in breast cancer cells using Apo-BrdU-Red in situ DNA fragmentation assay kit (BioVision, Milpitas, CA, USA) as described earlier. 26 Flow cytometric analysis was carried out using BD FACSVerse (BD Biosciences). The experiments were carried out in triplicates three independent times.
M30 assay
Caspase activation assay was performed using M30 antibody (Roche Diagnostics GmbH, Mannheim, Germany) and anti-mouse FITC (Jackson ImmunoResearch Laboratories) as described previously. 28 Data acquisition and analysis were examined using BD FACSVerse (BD Biosciences).
Cell migration and invasion assay
To study the role of AKAP4 in cellular motility, migration, and invasion, assays were carried in BD Biosciences Boyden chamber (Becton Dickinson Labware, Bedford, MA, USA) as described earlier. 26
ROS detection assay and N-acetyl cysteine treatment
ROS detection assay was performed in MDA-MB-231 and MCF-7 cells in AKAP4 shRNA transfected breast cancer cells using CellROX Deep Red (Thermo Fisher Scientific, Waltham, MA, USA) as described earlier. 26 The results were analyzed using BD FACSVerse, and the analysis was done using BD FAC Suite software (BD Biosciences). Quenching of ROS generation by antioxidant N-acetyl cysteine (NAC; Sigma-Aldrich) was carried out as described earlier. 26
Xenograft studies and immunohistochemistry studies
Effect of knockdown of AKAP4 on MDA-MB-231 tumor growth was examined in severe combined immunodeficiency (SCID) mice after obtaining ethical clearance from Institute Animal Ethics Committee (IAEC No. 360/14) as described earlier. 28 Immunohistochemistry (IHC) studies for various molecules involved in different pathways were carried out as described earlier. 28 Images were captured using Nikon Eclipse E400 microscope (Nikon, Fukuoka, Japan).
Statistical analysis
Student’s t-test (two-tailed) was performed to analyze the data for all in vitro and in vivo assays using SPSS 20.0 statistical software package (SPSS Inc., Chicago, USA). Data were expressed as mean ± standard error of the mean. The experiments were carried out in triplicates three independent times in in vitro assays. A value of p < 0.05 was considered statistically significant.
Results
AKAP4 is involved in breast cancer cell growth
AKPA4 protein localization was examined in MDA-MB-231 (basal TNBC ER− PR− HER2−) and MCF-7 (luminal breast adenocarcinoma ER+ PR+ HER2−) by confocal microscopy. Confocal images showed AKAP4 protein expression predominantly in cytoplasm and co-localized with various organelles such as ER, Golgi bodies, and mitochondria, whereas no AKAP4 co-localization was detected with nuclear envelope (Figure 1(a)).

AKAP4 protein expression is involved in cellular growth. (a) Confocal images depicting AKAP4 protein expression predominantly in cytoplasm. Co-localization (orange-yellowish staining) with endoplasmic reticulum, Golgi bodies, and mitochondria has been shown as orange-yellowish staining. No co-localization was detected with nuclear envelope. (b) Bar diagram depicting significant cell number reduction at 24, 48, and 72 h in AKAP4 downregulated MDA-MB-231 and MCF-7 cells compared to NC shRNA transfected cells. (c) Bar diagram depicting reduced cellular viability at 24, 48, and 72 h by MTT assay in AKAP4 downregulated MDA-MB-231 and MCF-7 cells compared to NC shRNA. (d) Flow cytometric analysis of cell cycle in AKAP4 shRNA2 or shRNA3 transfected MDA-MB-231 and MCF-7 cells compared to NC shRNA. Histogram and bar diagram depict the majority of cell population (stained with propidium iodide (PI)) accumulation in G0/G1 phase of cell cycle in shRNA2 or shRNA3 transfected MDA-MB-231 (58.85%: NC shRNA, 66.91%: shRNA2, and 66.3%: shRNA3) and in shRNA2 or shRNA3 transfected MCF-7 (62.49%: NC shRNA, 68.27%: shRNA2, and 68.02%: shRNA3) compared to NC shRNA (M1: G0/G1, M2: S, and M3: G2/M phase). (e) Western blot analysis depicts downregulation of CDK1, CDK2, CDK4, CDK6, cyclin A1, cyclin B1, cyclin D1, cyclin E, and PCNA and upregulation of p21 in AKAP4 shRNA2 or shRNA3 MDA-MB-231 and MCF-7 transfected cells compared to NC shRNA. β-actin was used as internal loading control. All experiments were carried out in triplicates three independent times. Data are represented as mean ± standard error of the mean (*p < 0.05, **p < 0.01, and ***p < 0.001; original magnification ×630, objective ×63).
In order to examine the knockdown efficiency of AKAP4, three shRNA targets shRNA1, shRNA2, or shRNA3 against AKAP4 gene were used in MDA-MB-231 and in MCF-7 cells. qPCR analysis showed significant reduction (p < 0.001) of relative messenger RNA (mRNA) expression of AKAP4 in shRNA1, shRNA2, and shRNA3 transfected (78%, 83%, and 87%, respectively) MDA-MB-231 cells compared to NC shRNA (Supplementary Figure S1a). We also observed significant reduction (p < 0.001) of AKAP4 mRNA in MCF-7 cells when transfected with shRNA1, shRNA2, and shRNA3 (72%, 82%, and 77%, respectively) compared to NC shRNA (Supplementary Figure S1a). Subsequently, all in vitro and in vivo studies were carried out based on gene silencing efficiency of shRNA2 and shRNA3. Western blot analysis revealed downregulation of AKAP4 protein in both shRNA2 and shRNA3 transfected MDA-MB-231 and MCF-7 cells compared to NC shRNA (Supplementary Figure S1b). We also observed significant reduction (p < 0.001) in cellular growth and cell viability in MDA-MB-231 and MCF-7 cells (Figure 1(b) and (c)) when transfected with shRNA2 or shRNA3 compared to NC shRNA. In addition, knockdown of AKAP4 resulted in significant reduction (p < 0.001) in colony forming abilities in MDA-MB-231 cells and MCF-7 (Supplementary Figure S1c). Cell cycle analysis in AKAP4 downregulated MDA-MB-231 and MCF-7 cells showed G0/G1 arrest phase (Figure 1(d)). Subsequently, we examined the effect of knockdown of AKAP4 on various molecules involved in different phases of cell cycle. Decreased expression of cyclins (cyclin A1, cyclin B1, cyclin D1, and cyclin E), CDKs (CDK1, CDK2, CDK4, and CDK6), and PCNA was observed in AKAP4 downregulated cancer cells by Western blot. Interestingly, the CDK inhibitor p21 expression was higher in both MDA-MB-231 and MCF-7 cells after AKAP4 shRNA (shRNA2 or shRNA3) transfection (Figure 1(e)).
Knockdown of AKAP4 enhances the cytotoxic effect of paclitaxel in breast cancer cells
To examine the effect of combination of AKAP4 downregulation and paclitaxel on cellular growth and viability in MDA-MB-231, MTT assay was carried out. We observed that combination treatment was significantly more cytotoxic (p < 0.001) than paclitaxel alone (Figure 2(a) and (b)). We further assessed AKAP4 shRNA and paclitaxel drug combination in non-constant ratio in MDA-MB-231 cells using the median-effect analysis method. Different concentrations of paclitaxel and AKAP4 shRNA at 48 h showed significant decrease in cell viability in a dose-dependent manner in MDA-MB-231 cells (Figure 2(c)). The IC50 values were 3.8, 3.78, and 14.17 μg/mL for AKAP4 shRNA2, AKAP4 shRNA3, and paclitaxel treatment, respectively. Interestingly, single or combinatorial treatment studies revealed that the values of CI were <1 (Figure 2(c) and Table 1), indicating that the combination of two treatment resulted in a synergistic effect.

Combinatorial effect of paclitaxel and AKAP4 shRNA results in enhanced cytotoxicity. (a) Bar diagram depicting significant reduction in MDA-MB-231 cell number at 24, 48, and 72 h with paclitaxel drug treatment alone or in combination with AKAP4 shRNA2 or AKAP4 shRNA 3. (b) Bar diagram depicting significant reduction in cellular viability of MDA-MB-231 cells at 24, 48, and 72 h with paclitaxel drug treatment alone or in combination with AKAP4 shRNA2 or AKAP4 shRNA3. (c) Dose–effect curve of combinatorial treatment with paclitaxel and AKAP4 shRNA2 by the median-effect method demonstrated that the values of combination index (CI) were <1 (d) Dose–effect curve of combinatorial treatment with paclitaxel and AKAP4 shRNA3 by the median-effect method demonstrated that the values of CI were <1. All experiments were carried out in triplicates three independent times. Data are represented as mean ± standard error of the mean (***p < 0.001).
The combination index (CI) values for the AKAP4 shRNA and paclitaxel in MDA-MB-231 cells.
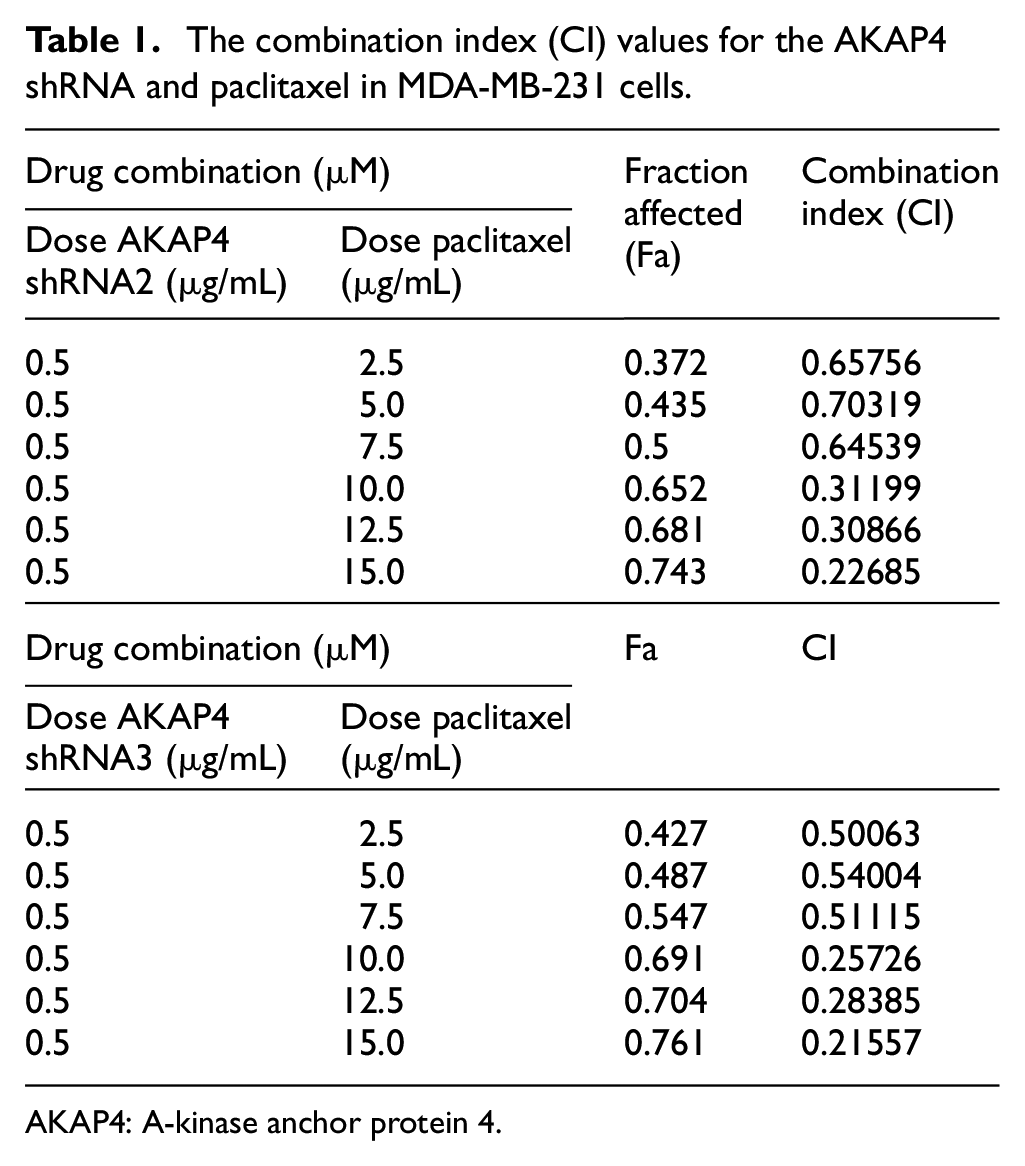
AKAP4: A-kinase anchor protein 4.
Knockdown of AKAP4 initiates apoptosis
Biochemical changes during the apoptosis process due to AKAP4 downregulation were subsequently assessed in both breast cancer cells. The onset of apoptosis was examined by Annexin V-PerCP-Cy5-5-A assay. Flow cytometric analysis of MDA-MB-231 cells revealed 31.15% and 42% annexin V expression in cells transfected with AKAP4 shRNA2 or shRNA3, respectively, compared to 6% with NC shRNA. Similarly, MCF-7 cells showed 34.50% and 52.26% of annexin V expression in cells transfected with AKAP4 shRNA2 or shRNA3, respectively, compared to 1.37% of NC shRNA (Figure 3(a) and Supplementary Figure S2a). Furthermore, to strengthen our findings, DNA fragmentation using TUNEL assay was carried, which showed 42.43% and 31.8% of BrdU-positive MDA-MB-231 cells when transfected with AKAP4 shRNA2 or shRNA3, respectively, compared to 1.02% with NC shRNA. Similarly, higher percentage of BrdU positivity was observed in MCF-7 cells transfected with AKAP4 shRNA2 (54.19%) or shRNA3 (51.8%) compared to NC shRNA (2.56%) (Figure 3(b) and Supplementary Figure S2b). Our results show that onset of apoptosis was found in higher percentage of MCF-7 cells compared to MDA-MBA-231 cells as determined by annexin V assay and TUNEL assay. However, significant onset of apoptosis was also observed in AKAP4 downregulated MDA-MB-231 cells. These observations may suggest that it is quite likely that TNBC cells are more aggressive and difficult to treat. Onset of apoptosis after initiation of effector caspases results in cleavage of CK18 into proteolytic fragments exhibiting neoepitopes (NE) at cleavage site that can be detected by M30 assay. Therefore, M30 assay was used to examine the effect of knockdown of AKAP4 in breast cancer cells that revealed higher M30 epitope expression in MDA-MB-231 cells transfected with AKAP4 shRNA2 (53.91%) or shRNA3 (40.63%) compared to NC shRNA (1.83%). Similarly, higher expression of M30 epitope was also observed in MCF-7 cells when transfected with AKAP4 shRNA2 (33.05%) or shRNA3 (25.02%) compared to NC shRNA (2.26%; Figure 3(c) and Supplementary Figure S2c). Furthermore, employing SEM, phenotypic changes associated with apoptosis in AKAP4 downregulated cells were captured. SEM images revealed membrane blebbing and presence of distorted structures in MDA-MB-231 and MCF-7 cells after 12 h of treatment with shRNA2 or shRNA3 which continued to increase by 48 h (Figure 3(d)). As expected, no changes were observed with NC shRNA treatment. We further investigated various molecules involved in the process of apoptosis in AKAP4 downregulated cells. Western blot analysis revealed upregulation of pro-apoptotic molecules such as BAX, BAD, BID, cytochrome-C, PUMA, APAF1, and caspases in AKAP4 downregulated breast cancer cells (Figure 3(e)). As expected, AKAP4 downregulation also resulted in downregulation of anti-apoptotic molecules such as BCL-2, Bcl-xL, and MCL-1 in both the breast cancer cells (Figure 3(e)). Our data suggest that AKAP4 plays an important role in cellular growth, viability, and apoptosis.

Knockdown of AKAP4 results in initiation of apoptosis. (a) Histogram depicts flow cytometric analysis of effect of knockdown of AKAP4 on apoptosis onset by annexin V assay which revealed increased phosphatidylserine surface expression in AKAP4 shRNA2 or shRNA3 MDA-MB-231 and MCF-7 transfected cells, as shown by the shift on x-axis, compared with NC shRNA. (b) Histogram depicts flow cytometric analysis of effect of knockdown of AKAP4 on DNA damage by TUNEL assay which revealed enhanced DNA fragmentation in AKAP4 shRNA2 or shRNA3 MDA-MB-231 and MCF-7 transfected cells as shown by the shift on x-axis, compared with NC shRNA. (c) Histogram depicts flow cytometric analysis of effect of knockdown of AKAP4 on caspase activation by M30 assay in AKAP4 shRNA2 or shRNA3 MDA-MB-231 and MCF-7 transfected cells as shown by the shift on x-axis, compared with NC shRNA. (d) Representative images show changes due to apoptosis in AKAP4 downregulated MDA-MB-231 and MCF-7 cells. Membrane blebbing and distorted structures in cells were observed after 24 and 48 h post-transfection with AKAP4 shRNA2 or shRNA3 compared to NC shRNA (magnification ×6000, WD = 6 mm, EHT = 20.00 kV (e) Western blot analysis shows that AKAP4 downregulation resulted in upregulation of BAX, BAD, BID, cytochrome-C, PUMA, APAF1, and caspases, whereas downregulation of BCL-2, Bcl-xL, and MCL-1 in AKAP4 shRNA2 or shRNA3 transfected MDA-MB-231 and MCF-7 cells compared to NC shRNA. β-actin was used as internal loading control.
Upregulation of CHOP and NOXA in AKAP4 knockdown breast cancer cells
CHOP has been shown to act as ER stress effector that targets several apoptotic genes, including NOXA. As a result of upregulation of apoptotic genes and NOXA, onset of apoptosis occurs, which leads to cell death. Therefore, we examined the expression of CHOP at protein level by Western blotting. Our data showed that AKAP4 downregulated MDA-MB-231 cells resulted in upregulation of CHOP protein expression along with NOXA (Figure 4(a)). Interestingly, when we knock down CHOP in MDA-MB-231 cells by shRNA approach, it did not alter AKAP4 expression and no NOXA expression was detected (Figure 4(b)). To further confirm the relationship between CHOP, NOXA, and AKAP4 induced apoptosis, we carried out co-transfection studies in TNBC cells. Subsequently, when TNBC cells were co-transfected with AKAP4 shRNA and CHOP shRNA, we observed the downregulation of CHOP and cleaved PARP protein expression (Figure 4(c)). Furthermore, we also documented the upregulation of NOXA in time-dependent experiment along with cleaved PARP in AKAP4 downregulated cells (Figure 4(d)). Similarly, when we knock down NOXA by shRNA approach in MDA-MB-231 cells, it did not result in upregulation of CHOP or alter AKAP4 expression (Figure 4(e)). When we co-transfected cells with AKAP4 shRNA and NOXA shRNA, we found downregulation of NOXA and cleaved PARP expression (Figure 4(f)). Taken together, our findings demonstrate that NOXA/CHOP are unregulated in AKAP4 knockdown cells and are involved in the initiation of apoptosis.

Downregulation of AKAP4 induces apoptosis mediated through NOXA/CHOP. (a) AKAP4 downregulation results in upregulation of CHOP and NOXA expression in MDA-MB-231 cells. (b) Knockdown of CHOP did not alter the AKAP4 expression, and as expected no expression of CHOP or NOXA was detected in MDA-MB-231 cells. (c) Expression of CHOP and cleaved PARP was decreased when cells were co-transfected with AKAP4 shRNA and CHOP shRNA compared to AKAP4 shRNA transfected cells alone. (d) Knockdown of AKAP4 results in upregulation of NOXA and cleaved PARP at different time points as analyzed by Western bloating. (e) Knockdown of NOXA did not alter the AKAP4 expression, and as expected no expression of NOXA and CHOP was detected in MDA-MB-231 cells. (f) Expression of NOXA and cleaved PARP was also decreased when MDA-MB-231 cells were co-transfected with AKAP4 shRNA and NOXA shRNA compared to AKAP4 shRNA transfected cells alone. (g) Bar diagram represents significant increase in ROS generation in mean fluorescence intensity (MFI) in AKAP4 shRNA2 or AKAP4 shRNA3 transfected MDA-MB-231 and MCF-7 cells compared to NC shRNA. (h) MDA-MB-231 cells transfected with AKAP4 shRNA resulted in high expression of NOXA and cleaved PARP. However, reduced expression of NOXA and cleaved PARP was detected when MDA-MB-231 cells were pretreated with NAC (4 µM) for 4 h and subsequently transfected with AKAP4 shRNA3.
Upregulation of CHOP/NOXA causes excessive ROS generation
In order to investigate the downregulation of AKAP4 based apoptosis, we explored the potential involvement of ROS production in MDA-MB-231 and MCF-7 cells. Our results showed that MDA-MB-231 and MCF-7 cells transfected with AKAP4 shRNA2 resulted in 2.06 fold and 1.64 fold and with AKAP4 shRNA3 resulted in 3.4 fold and 1.69 fold increased ROS generation compared to NC shRNA (Figure 4(g)). To test whether the potentiating effect of AKAP4 downregulated TNBC death was mediated via ROS, we used antioxidant N-acetyl cysteine (NAC) that is a widely used ROS scavenger. As a result of NAC treatment, the AKAP4 downregulated cells revealed decreased level of cleaved PARP (Figure 4(h)). We also found that downregulation of AKAP4 results in increased levels of NOXA. Interestingly, NAC pre-treatment in TNBC cells resulted in reduced expression of NOXA (Figure 4(h)), indicating that ROS does act on apoptotic genes.
Downregulation of AKAP4 inhibits the migrating and invasive potential of breast cancer cells
Increased cellular motility and invasive abilities of cancer cells are considered as the hallmark of metastasis. In this context, the migratory ability of breast cancer cells after AKAP4 shRNA transfection was examined in MDA-MB-231 cells. Migration assay revealed significant reduction in cells transfected with AKAP4 shRNA2 (41.75%; p < 0.001) or shRNA3 (44.09%; p < 0.001) compared to NC shRNA (Figure 5(a)) in MDA-MB-231 cells. Significant loss of invasive ability into Transwell through Matrigel was also observed in MDA-MB-231 cells transfected with AKAP4 shRNA2 (65.76%; p < 0.001) or shRNA3 (64.09%; p < 0.001) in contrast to NC shRNA (Figure 5(b)). EMT is considered an important developmental process for cancer cells to gain migrating and invasive properties. Downregulation of AKAP4 resulted in decreased expression of SLUG, TWIST, Vimentin, N-cadherin, and P-cadherin in both breast cancer cells. As expected, knockdown of AKAP4 revealed increased E-cadherin expression (Figure 5(c)), supporting our observation of reduced migration and invasive abilities of cancer cells.

Knockdown of AKAP4 reduces migration and invasion abilities of breast cancer cells. (a) Phase contrast image and bar diagram depict significant reduction in cell migration into Transwell through insert membrane after AKAP4 downregulation by shRNA2 or shRNA3 in MDA-MB-231 cells compared to NC shRNA. (b) Phase contrast image and bar diagram depict significant reduction in cell invasion into Transwell through Matrigel membrane after AKAP4 downregulation by shRNA2 or shRNA3 in MDA-MB-231 cells compared to NC shRNA. (c) Western blot analysis of various EMT molecules depicting downregulation of SLUG, TWIST, Vimentin, N-cadherin, and P-cadherin, whereas upregulation of E-cadherin in AKAP4 shRNA2 or shRNA3 MDA-MB-231 and MCF-7 transfected cells compared to NC shRNA. β-actin was used as internal loading control. All experiments were carried out in triplicates three independent times. Data are represented as mean ± standard error of the mean (***p < 0.001).
Knockdown of AKAP4 reduces tumor growth
Our in vitro studies on AKAP4 downregulation showed reduction in cellular proliferation in TNBC cells. Hence, in vivo studies were carried out using triple-negative MD-MBA-231 cells. Knockdown of AKPA4 resulted in significant reduction (p < 0.01) of tumor size and volume compared to NC shRNA treated group (Figure 6(a) and (b)). Also, tumor lysate from mice treated with AKAP4 shRNA3 showed reduced AKAP4 and PCNA protein expression (Figure 6(c)). Furthermore, IHC analysis of paraffin-embedded MDA-MB-231 xenograft tumor sections showed reduced immunoreactivity of AKAP4 and PCNA in AKAP4 shRNA3 treated tumor sections (Figure 6(d)). Next, we checked the expression of cell cycle regulators in xenograft tumor sections employing IHC. Our data revealed reduced immunoreactivity of cyclins (cyclin D1 and cyclin B1) and CDKs (CDK1, CDK4, and CDK6) in AKAP4 shRNA3 treated group. As expected, upregulation of CDK inhibitor p21 showed increased immunoreactivity in AKAP4 downregulated tumor sections (Figure 6(e)).

Knockdown of AKAP4 reduces tumor growth of MDA-MB-231 xenograft. (a) Representative image shows reduced tumor size dissected from animals when treated with AKAP4 shRNA3 compared to NC shRNA treated tumor. (b) Representative graph depicts the reduction in tumor size in AKAP4 shRNA3 treated mice compared to NC shRNA treated mice. (c) Western blot analysis of tumor lysate indicates reduced expression of AKAP4 and PCNA in shRNA3 treated mice compared to NC shRNA treated mice. β-actin was used as internal loading control. (d) IHC analyses of tumor section depict reduced expression of AKAP4 and PCNA in AKAP4 shRNA3 treated mice compared to NC shRNA treated mice. Cell cytostructure of tumor stained with hematoxylin and eosin. (e) IHC analyses of cell cycle molecules in tumor treated with AKAP4 shRNA3 depict reduced immunoreactivity of cyclin D1, cyclin B1, CDK1, CDK4, and CDK6 and enhanced immunoreactivity of p21 compared to NC shRNA treated tumor. (f) IHC analyses of various molecules involved in apoptotic pathways in AKAP4 shRNA3 treated mice show increased immunoreactivity of pro-apoptotic molecules such as BAD, APAF1, PUMA, cytochrome-C and caspase 3, whereas decreased immunoreactivity of anti-apoptotic molecules Bcl-xL compared to NC shRNA treated mice. (g) IHC analyses of various molecules involved in epithelial–mesenchymal transition (EMT) in mice treated with AKAP4 shRNA3 depict decreased immunoreactivity of P-cadherin, N-cadherin, Vimentin, and TWIST, whereas increased immunoreactivity of E-cadherin compared to NC shRNA treated mice. All experiments were carried out in triplicates three independent times. Data are represented as mean ± standard error of the mean (**p < 0.01; original magnification ×200, objective ×20).
We further investigated the effect of AKAP4 downregulation on pro-apoptotic molecules and anti-apoptotic molecule in tumor section by employing IHC. We observed an increased immunoreactivity of pro-apoptotic molecules (BAD, APAF-1, PUMA, and cytochrome-C) in AKAP4 downregulated tumor sections compared to the NC shRNA group. Interestingly, upregulation of caspase 3 was also observed in AKAP4 downregulated tumor sections. In addition, there was downregulation of anti-apoptotic molecule (Bcl-xL) in the AKAP4 downregulated tumor sections (Figure 6(f)). EMT molecules were also examined in xenograft tumor sections. We found decreased immunoreactivity of P-cadherin, N-cadherin, Vimentin, and TWIST in AKAP4 downregulated tumor sections compared to the NC shRNA group. As expected, increased reactivity of E-cadherin was observed in AKAP4 shRNA3 treated tumor sections (Figure 6(g)). Our in vivo studies indicate that reduced AKAP4 expression does result in reduced tumor growth.
Discussion
TNBC represents a heterogeneous subtype with poor prognosis. 31 To date, cytotoxic chemotherapy remains as the standard systemic treatment; however, no effective specific targeted therapy is readily available. In recent years, very few studies have shown some promise in preliminary clinical studies including PARP inhibitors, epidermal growth factor receptor (EGFR) inhibitors, and immunotherapies; hence, in-depth investigations are critically needed in the identification of novel candidate molecules for therapeutic options for TNBC treatment.32–34 Recently, we demonstrated that AKAP4 was associated with different clinico-pathological features of breast cancer. 24 Therefore, in the present investigation, we examined the involvement of AKAP4 in TNBC cell growth and showed that AKAP4 downregulation caused upregulation of CHOP and NOXA, generation of ROS, DNA damage, cell cycle arrest, initiated apoptosis, inhibited cellular motility in vitro, and reduced tumor growth in triple-negative MDA-MB-231 xenograft studies. Our data indicated enhanced cytotoxic effect of paclitaxel in AKAP4 downregulated TNBC cells. Therefore, AKAP4 may be potential target and be used in combination with paclitaxel for TNBC management and warrants future studies.
Most importantly, one of the features of cancer cell is uncontrolled cellular proliferation due to loss of cell cycle control. 35 Interestingly, knockdown of AKAP4 resulted in reduced cellular proliferation and colony forming ability in TNBC cells. Recent studies on knockdown of Rho GTPase activating protein 9 (ARHGAP9) also revealed decreased clonogenic potential in TNBC cells. 36 This is in line with our earlier studies where knockdown of AKAP4 led to decreased colony-forming ability in cervical cancer cells, 25 ovarian cancer cells, 26 and colorectal cancer cells. 28
Despite the good initial response to chemotherapy, TNBC tumors develop resistance to various chemotherapeutic regimes. Moreover, the prognosis of TNBC patients remains poor compared to non-TNBC. Our study showed that AKAP4 knockdown in fact enhances the cytotoxic effect of paclitaxel in TNBC cells. Our recent study also revealed similar observation that showed increased cytotoxicity with paclitaxel in SPAG9 knockdown ovarian cancer cells. 30 Yet another study also showed that knockdown of CDK11 increased ovarian cancer cell death when treated along with paclitaxel. 37 However, increased paclitaxel cytotoxicity in AKAP4 downregulated TNBC cells requires in-depth understanding of molecular pathways and warrants further studies.
The cell cycle process is carefully controlled, and hence loss of cell cycle control may eventually lead to tumor development. 38 Intrigued by AKAP4 involvement in cellular growth, we observed that AKAP4 downregulation caused the cell cycle arrest in the G0/G1 phase. Recently, we demonstrated knockdown of AKAP4 that resulted in s-phase arrest in ovarian cancer cells. 26 The arrest in different stages of cell cycle process may be due to different origins of cancer cell type and may involve in different molecular pathways. 39 It has been demonstrated that the initiation of various phases of cell cycle is tightly regulated by cyclins and CDKs. 39 Interestingly, our study demonstrated that knockdown of AKAP4 resulted in reduced expression of cyclin B1, cyclin D1, and cyclin E along with CDK1, CDK4, and CDK6, which might contribute toward G0/G1 arrest. Furthermore, as a result of downregulation of cyclins and CDKs, we also observed reduced cellular and tumor growth. Earlier studies in yeast and mammalian cells have demonstrated that cyclin D1 and CDK4 support the transition of cell from the G0/G1 phase to S phase. 38 In addition, in breast cancer cells, downregulation of KIF26B showed reduced expression of cyclin D1 and cyclin E along with their CDK partners, leading to inhibited cellular proliferation. 40 Yet another study on TASK-3 potassium channel revealed that knockdown of TASK-3 in MDA-MB-231 cells resulted in G0/G1 cell cycle arrest as a result of upregulation of CDK inhibitors p21 and p27. 41 Interestingly, our findings also demonstrated that knockdown of AKAP4 caused increased expression of cell cycle inhibitor p21 that resulted in G0/G1 cell cycle arrest. We further demonstrated that knockdown of AKAP4 results in apoptosis. Recently, our group showed increased apoptosis in AKAP4 downregulated ovarian cancer cells. 26 Our group also reported another study on SPAG9 and showed that knockdown of SPAG9 enhanced programmed cell death in TNBC cells. 42 Our data indicate that AKAP4 promotes cellular growth. Moreover, knockdown of AKAP4 causes cell cycle arrest, apoptosis, and hence cell death.
Excessive ROS generation is known to cause DNA damage and various proteins that lead to apoptosis.43,44 One of the major findings of our study is that AKAP4 knockdown leads to increased ROS generation and TNBC cell death mediated via CHOP/NOXA axis. Recently, we have established the correlation between AKAP4 expression and ROS generation in ovarian cancer. 26 In this study, we report high expression of NOXA as a result of ROS generation. It is documented that NOXA is activated by various factors, and ROS is one of the causative factors. 45 It has also been reported that CHOP transcription factor regulates NOXA activation. 46 Interestingly, we also found that knockdown of AKAP4 induced ROS generation as a result of upregulation of CHOP and NOXA. Thus, these findings together suggest that AKAP4 knockdown induces apoptosis as a result of increased ROS generation mediated by NOXA/CHOP axis.
The majority of cancer associated deaths are due to primary tumor metastasis. 47 Various molecules/targets have been demonstrated to be involved in cellular motility; however, none of these targets have been used in clinical practice for TNBC management. 48 Previously, AKAP4 has been shown to be associated with increased metastasis in in vitro ovarian cancer cells and colorectal cancer cells, respectively.26,28 Our data suggest that as a result of downregulation of EMT molecules, we show reduced migration and invasive abilities in AKAP4 downregulated TNBC cells. In this regard, our data are consistent with recent findings in cancer/testis antigen family 45 member (CT45A1), wherein CT45A1 overexpression resulted in increased migration and invasion abilities in breast cancer cells. 48 It is well established that cancer cells acquire migratory and invading abilities due to loss of E-cadherin expression. 49 Our data further reveal that AKAP4 downregulated cells demonstrated increased expression of E-cadherin in triple-negative MDA-MB-231 xenograft tumor sections, indicating the possible role of AKAP4 in the metastatic potential of TNBC cells.
Conclusion
In summary, we have demonstrated that AKAP4 plays an important role in TNBC cell growth. Our findings suggest that AKAP4 may be a potential therapeutic target and may have clinical application for TNBC management. Further studies are warranted.
Supplemental Material
SUPLLEMENNTARY_MATERIAL – Supplemental material for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo
Supplemental material, SUPLLEMENNTARY_MATERIAL for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo by Nirmala Jagadish, Sonika Devi, Namita Gupta, Vitusha Suri and Anil Suri in Tumor Biology
Supplemental Material
SUPPLEMENTARY_FIGURE_S1 – Supplemental material for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo
Supplemental material, SUPPLEMENTARY_FIGURE_S1 for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo by Nirmala Jagadish, Sonika Devi, Namita Gupta, Vitusha Suri and Anil Suri in Tumor Biology
Supplemental Material
SUPPLEMENTARY_FIGURE_S2 – Supplemental material for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo
Supplemental material, SUPPLEMENTARY_FIGURE_S2 for Knockdown of A-kinase anchor protein 4 inhibits proliferation of triple-negative breast cancer cells in vitro and in vivo by Nirmala Jagadish, Sonika Devi, Namita Gupta, Vitusha Suri and Anil Suri in Tumor Biology
Footnotes
Author’s contribution
N.J., S.D., N.G., and V.S. standardized and carried out experiments. N.J. and N.G. performed acquisition analysis and interpretation of data. N.G. and N.J. prepared figures and drafted the manuscript. A.S. developed the concept and designed the study. All authors read and approved the manuscript.
Availability of data and material
Information is included in section “Materials and methods.”
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Indo-UK Cancer Research Program (Grant No. BT/IN/UK/NII/2006), ICMR #5/10/CAR/4/2018-RBMCH, and NII-core funding, Department of Biotechnology, Government of India.
Institute Animal Ethics committee (IAEC) approval
IAEC approval has been obtained (IAEC no. 360/14). This information is included in section “Materials and methods.”
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.