
Abstract
Genome-wide association studies (GWAS) have identified genetic variants associated with an increased risk of developing breast cancer. However, the association of genetic variants and their associated genes with the most aggressive subset of breast cancer, the triple-negative breast cancer (TNBC), remains a central puzzle in molecular epidemiology. The objective of this study was to determine whether genes containing single nucleotide polymorphisms (SNPs) associated with an increased risk of developing breast cancer are connected to and could stratify different subtypes of TNBC. Additionally, we sought to identify molecular pathways and networks involved in TNBC. We performed integrative genomics analysis, combining information from GWAS studies involving over 400,000 cases and over 400,000 controls, with gene expression data derived from 124 breast cancer patients classified as TNBC (at the time of diagnosis) and 142 cancer-free controls. Analysis of GWAS reports produced 500 SNPs mapped to 188 genes. We identified a signature of 159 functionally related SNP-containing genes which were significantly (
Introduction
Recent advances in high-throughput genotyping and reduction in genotyping costs have made it possible to identify genetic variants associated with an increased risk of breast cancer possible by using genome-wide association studies (GWAS).1–3 These findings are providing valuable clues about the genetic susceptibility landscape of breast cancer. However, the association of genetic variants and their associated genes with the most aggressive subset of breast cancer, the triple negative breast cancer (TNBC) remains a central puzzle in molecular epidemiology. Currently, there is a substantial gap between single nucleotide polymorphism (SNP) associations from GWAS and understanding how susceptibility loci contribute to the TNBC phenotype.
TNBC–-tumors that do not express estrogen receptors, progesterone receptors, or HER2–-are typically high-grade duct carcinomas, although low-grade tumors do occur.4,5 They represent an important clinical challenge because these cancers do not respond to endocrine therapy or other available targeted therapies.6,7 TNBC is significantly more aggressive than other subtypes of breast cancer and disproportionately affects younger premenopausal women, with a higher mortality rate among African-American women. 8 Patients with TNBC have a significantly increased risk of relapse and shorter survival rate than patients affected with tumors of other molecular subtypes. 5 In fact, although TNBC accounts for a relatively small proportion of breast cancer cases–-about 15% to 20% of all breast cancers diagnosed in the general US population and about 30% in the African-American population-it is responsible for a disproportionate number of breast cancer deaths.9,10 Additionally, a lower proportion of TNBCs are discovered by mammographic screening, possibly due partly to the age distribution of patients afflicted by this disease.11,12
To date, there have been fewer advances in the treatment of TNBC compared to other subtypes of cancer. Although these tumors respond to conventional chemotherapy, which is toxic and affects a wide range of dividing cells, the approach has met with mixed success. 5 TNBC is often aggressive and highly resistant to chemotherapy. 5 TNBC relapses more frequently than hormone receptor-positive, luminal subtypes, and have a worse prognosis. 5 The five-year survival rate for TNBC is about 77%, compared to 93% for other types of breast cancer. 5 An important goal is therefore the identification of molecular markers to reliably identify high and low risk subsets of patients with TNBC, both for different treatment approaches and for the development of novel, more effective therapeutic strategies.
Over the last decade, transcription profiling using gene expression microarray technology has made possible the systematic molecular stratification of TNBC.8,13 Microarrays have also been used for the histopathological characterization of TNBC. 14 More recently, gene expression profiling has been used to identify triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. 15 However, while these primary analyses have made it possible to identify molecular signatures of TNBC, they have been unsuccessful in identifying which genes and pathways have causative roles as opposed to being consequences of the disease states. 2 Recently, several genetic variants associated with TNBC were reported.16,17 However, the reported genetic variants associated with TNBC–-which include rs2046210 (ESR1), rs12662670 (ESR1), rs3803662 (TOX3), rs999737 (RAD51L1), rs8170 (19p13.1), and rs8100241 (19p13.1)–-explain only a small fraction of genetic variation and have modest effects.16,17 In addition, results from these studies provide no information about the functional roles and the broader context in which the identified susceptibility loci operate leading to the TNBC phenotype.
Because genetic variants that are associated with an increased risk for breast cancer do not lead directly to the disease but instead act on intermediate molecular phenotypes (gene expression), a more optimal approach is to combine GWAS information with gene expression data. Integration of GWAS information with gene expression data provides a unified and powerful approach for identifying potential biomarkers and biological pathways dysregulated in TNBC. The objective of this study was to determine whether genes containing single nucleotide polymorphisms (SNPs) associated with an increased risk of developing breast cancer are both associated with TNBC and could stratify TNBC. A second but equally important objective was to identify molecular pathways and networks enriched for SNPs, which are dysregulated in TNBC. We hypothesized that genes containing SNPs (herein called genetic variants) associated with an increased risk of developing breast cancer are associated with TNBC. We further hypothesized that genes containing genetic variants associated with TNBC are functionally related and interact with each other in biological pathways and networks associated with TNBC. We tested these hypotheses using an integrative genomics approach which combines GWAS information with publicly available gene expression data derived from both breast cancer patients and cancer-free controls. The analysis strategy assumes a gene-centric approach in which the genes containing SNPs are treated as the units of association, with gene expression data derived from patients classified as TNBC at the time of diagnosis acting as the intermediate phenotype. Throughout this report we have used the terms genetic variants and SNPs interchangeably.
Material and Methods
Source of Snp Data
Our methods for data collection were based on guidelines proposed by the Human Genome Epidemiology network for the systematic review of genetic association studies; our methods follow the PRISMA guidelines.18–22 We have previously reported sources of SNP data, including references. 2 Here we provide a brief but detailed description of the methods used in data collection. We mined SNP data and gene information from published reports of GWAS on breast cancer. GWAS were eligible to be included if they met 4 criteria. First, the study design had to be a case-control, cohort or a cross-sectional association study conducted on unrelated individuals in human populations. Second, the study examined the association between breast cancer and the polymorphic phenotype, had a sample case size of greater than 500 subjects and greater than 500 controls, and provided sufficient information such that genotype frequencies for both breast cancer cases and controls could be determined without ambiguity. Third, breast cancer must have been diagnosed by pathological or histological examination. Fourth, publications must have been in peer-review journals or online and published in English on or before June 2012. Only studies published as full-length articles or letters in English language peer-reviewed journals were included in the analysis.
To identify all relevant publications, we used two strategies. First, we queried PubMed with terms (breast cancer, GWAS, GWA, WGAS, WGA, genomewide, genomewide, whole genome, and all terms + association or + scan) in combination with breast cancer to find all the GWAS published before June 2012. This study yielded 100 publications, which were screened by title, abstract, and full text review in order to identify studies that met our eligibility criteria. The data was manually extracted from both the reported GWAS and the supplementary data in websites accompanying those studies. It was then summarized in a consistent manner.
2
When a study included multiple ethnic populations, we picked the results of the model which adjusted for ethnicity. When possible we considered each subpopulation as an independent study. The search yielded 500 SNPs mapped to 188 genes from a population of over 400,000 cases and over 400,000 controls. The 100 genetic variants mapped to intergenic regions were not included in this analysis. Table D provides supplementary data on the 500 SNPs, including SNP ID, the genes they map to, assigned
To address publication bias, we catalogued all available SNPs that showed significant (
Gene Expression Data
We used publicly available gene expression data in our analysis. Data selection were based on the TNBC classification guidelines reported by Perou, which define TNBC as tumors which lack expression of estrogen receptor (ER), progesterone receptor (PR), and HER2. 8 A significant proportion of TNBC–-50% to 75%–-matches the basal-like molecular subtype. 8 However, TNBC is a heterogeneous disease entity encompassing other subtypes of cancer. Synonymous terms of this subtype include basal-type, basal-epithelial phenotype, basal breast cancer, and basaloid breast cancer. 24 Although most basal-like breast cancer is TNBC, not all cases are. In fact, even within basal-like/basal, molecular subtyping has revealed two subtypes,15,25 underscoring the complexity of the TNBC phenotype. Therefore, focusing on the basal-like subtype of TNBC alone might not be specific enough for biomarker discovery and identification of potential therapeutic targets. Because of inherent heterogeneity in the TNBC phenotype, we decided to include the other two subtypes, normal-like and nonluminal basal, both of which are classified as TNBC. 8 The gene expression data set used in this study comprised of 266 subjects, of which 124 were breast cancer patients. Of the 124 breast cancer patients, 29 were classified as normal-like, 20 were classified as basal-like, and 75 were classified as nonluminal basal as documented by data originators. 25 The remaining 142 subjects were cancer-free controls. These sample sizes were large enough to identify genes associated with TNBC with a statistical of power of 99%. We did not include the Claudin-low subtype 8 because we did not find a suitable data set that matched the other data sets used in this study; therefore, we acknowledge this weakness of the investigation.
All gene expression data was generated using the Affymetrix platform and the U133PLUS 2.0 Human Chip. The microarray data from these samples, including the raw probe-level hybridization intensities, were downloaded from the NCBI's Gene Expression Omnibus (GEO) database 26 under accession numbers, GSE21653, 25 and GSE7904, 27 respectively. Methods of sample collection, preparation, and processing have been fully described by the data originators.25,27 Data on the cancer-free controls GEO accession number GSE10780 has been fully described by the originators. 28 For each data set, the entries in the data matrix were average scaled difference expression values normalized using the RMA suite on a log scale (log2). Spiked control genes were removed from the data during preprocessing.
Data Analysis
We performed both unsupervised and supervised analyses followed by network modeling, visualization, and pathway prediction. The goal of this study was to determine whether genes containing SNPs associated with an increased risk of developing breast cancer are both associated with TNBC and could stratify TNBC. Additional, we aimed to identify gene regulatory networks and biological pathways enriched for SNPs which are dysregulated in TNBC. Therefore, as a first step in the analysis, we partitioned gene expression data into two subsets, a prioritized subset (ie, a data set of 188 genes containing SNPs associated with an increased risk of developing breast cancer) and a non-prioritized set (ie, a data set containing the remainder of the genes not identified by GWAS). Prioritization of SNP-containing genes was aimed at identifying the genes providing good evidence of association with TNBC, amongst a large pool of 188 SNP-containing genes identified by GWAS. The overarching goal was to maximize the yield and the biological relevance of further downstream screens, analysis, refinements, validation, functional analysis, pathway prediction, and network modeling focusing on the most promising candidates associated with TNBC.
We performed unsupervised analysis using hierarchical clustering on the prioritized data set to discern the patterns of gene expression profiles in TNBC for all the 188 genes containing SNPs associated with an increased risk of developing breast cancer. This unbiased class discovery approach included gene expression data on all 188 genes. Prior to clustering, the data was normalized using median normalization, standardized and centered. 29 Pairwise similarity of all the 188 genes was calculated as the Pearson correlation coefficient of the expression levels. The genes were then grouped by hierarchical clustering using the complete linkage method as implemented in GenePattern. 30
To obtain a more robust analysis on gene expression data and to identify significantly differentially-expressed SNP-containing genes that are both associated with TNBC and could stratify TNBC, we performed supervised analysis. The significant differences in gene expression profiles between cases and controls were tested using a
We then performed unsupervised analysis based on hierarchical clustering, using gene expression data on genes highly significantly (
To assess biological functional relationships, we performed additional analysis using the gene ontology (GO) information. 34 The GO Consortium has developed three separate categories (molecular function, biological process, and cellular component) to describe the attributes of gene products. Molecular function defines what a gene product does at the biochemical level without specifying where or when the event actually occurs or its broader context. Biological process describes the contribution of the gene product to the biological objective. Cellular component refers to where in the cell a gene product functions. Because our goal in this study was to gain biological insights about the broader context in which genetic variants associated with an increased risk of developing TNBC operate, we considered all three GO categories.
One of the limitations of GWAS as noted in this study is that the results of single-SNP GWAS analysis explain only a small fraction of variation. For example, in TNBC only a very small number of risk loci have been reported.16,17 This begs the question of where the missing variation is located. Realizing that there may be other key driver genes that act in concert with SNP-containing genes to produce the TNBC phenotypes, we performed additional analysis on the remainder of the data set (unprioritized data set) using supervised analysis. We then proceeded with an unsupervised analysis, as described earlier in this report. The data containing genes identified from this analysis was merged with the data set of SNP-containing genes significantly associated with TNBC. The combined data set was then subjected to unsupervised analysis using GenePattern 30 to identify co-expressed genes with similar expression profiles.
Finally, we performed pathway prediction, network modeling, and visualization using the Ingenuity System (IPA) program (http://www.ingenuity.com). 35 The goal was to identify biological pathways that are enriched by the genetic variants associated with breast cancer and which are also involved in TNBC. We hypothesized that genes containing SNPs associated with an increased risk for TNBC interact with each other and other genes within biological pathways. HUGO gene identifiers were mapped to networks available in the Ingenuity database and ranked by score. The score indicates the likelihood of the genes in a network being found together by random chance. Using a 99% confidence interval, scores of ≥3 are considered significant. Additional information, validation of predicted pathways, and identification of other downstream target genes was achieved through the literature and database mining module built in the Ingenuity System. This allowed identification of other functionally related genes not identified by GWAS. The distribution of the overall effect of SNPs in the pathway and replicated SNPs were calculated using the procedures developed by developed by Hicks et al. 2
Results
Patterns of Gene Expression Profiles for All Genes Containing Snps
As a first step, we performed exploratory analysis using unsupervised analysis to assess the patterns of expression profiles for all 188 genes containing 500 SNPs associated with an increased risk of developing breast cancer. Figure 1 shows the patterns of gene expression profiles for all 188 genes containing SNPs associated with an increased risk of developing breast cancer. SNP-containing genes exhibited patterns of expression profiles that were different from the controls (Fig. 1). Overall, patterns of gene expression profiles varied markedly, with basal-like subtypes showing more consistent patterns while basal and normal-like subtypes exhibited more variability and spurious patterns (Fig. 1). Patterns of expression profiles in the normal-like and non-luminal basal subtypes tended to be similar (Fig. 1). Patterns of expression profiles for some genes in the normal-like and basal subtypes were similar to cancer-free controls. The variability in patterns of gene expression was expected given the genetic and phenotypic heterogeneity inherent in TNBC.5,8,15,36 These results suggest that genetic susceptibility to TNBC could vary, posing challenges in identifying and stratifying breast cancer patients at risk. The spurious patterns in gene expression profiles suggest that some of the genes may not be associated with the TNBC subtypes under study (Fig. 1). Therefore, after this exploratory analysis, we performed more rigorous analyses and refinements as described in the methods section to identify genes that are both significantly associated with TNBC and could stratify TNBC.

Patterns of gene expression profiles for all the 188 genes containing SNPs associated with an increased risk of developing breast cancer, assessed in 142 controls and subtypes of TNBC (normal-like, N = 29; basal-like, N = 20; and non-luminal basal, N = 75) Tumors.
Association between Gwas Information and Tnbc
The primary objective of this investigation was to determine whether genes containing SNPs associated with an increased risk of developing breast cancer are associated with the most aggressive subset of breast cancer, TNBC. We hypothesized that genes containing SNPs associated with an increased risk of developing breast cancer significantly differ in their expression profiles between patients classified as TNBC and cancer-free controls. To test this hypothesis, we performed supervised analysis as described in the methods section of this report. The results showing estimates of
A comparison between normal-like, basal-like and basal subtypes with cancer-free controls produced 98 genes (
Estimates of

Interestingly, genes containing SNPs with small to moderate effects were found to be highly significantly associated with TNBC. This is a significant finding given that only a small number of statistically unimpeachable, common low-penetrance breast cancer susceptibility loci have been reported and confirmed in different populations and in TNBC.16,17 Of particular interest was the weak link or lack of association of both the FGFR2 gene with TNBC and the association of the
Overall, there was incomplete overlap in association between TNBC and cancer-free controls. This is attributable to heterogeneity in patterns of gene expression among the three types of TNBC studied. To assess overlap, we used a Venn diagram to delineate the overlapping genes. We sought to group the genes into those exhibiting significant association with all the three subtypes of TNBC, those associated with two subtypes, and those exhibiting subtype-specific association. Figure 2 shows the intersections of normal-like, basal-like, and basal subtypes. Overall, 119 genes were significantly associated with all three TNBC subtypes. Out of the remainder, 22 genes were significantly associated with the basal-like and basal subtypes only, 18 genes with basal and normal-like only, and 12 genes were significantly associated with basal-like and normal-like only (Fig. 2). Very few genes exhibited subtype-specific association (Fig. 2). Only three genes (not included in the Venn diagram) were not associated with any of the TNBC subtypes under study. These results are consistent with previous findings indicating that TNBC are heterogeneous and overlap is incomplete. 8 The heterogeneity and variability in the results suggests that genetic susceptibility in TNBC may not reflect a single disease, but rather a heterogeneous entity with some loci conferring risks to all subtypes, while others confer subtype-specific risks.
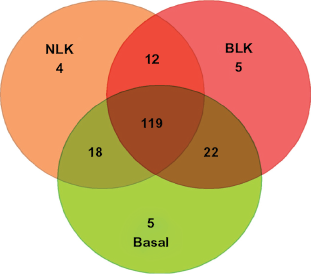
Venn diagram showing the numbers and distribution of genes significantly associated with TNBC subtypes under study.
Having observed heterogeneity and incomplete overlap in association between TNBC and cancer-free controls, we performed ANOVA among the three TNBC subtypes under study in order to quantify the extent of variation and to identify a set of genes which show significant group differences in expression profiles. We hypothesized that gene expression profiles significantly vary within and across the three TNBC subtypes under study, and that a subset of genes contribute to this significant variation. Estimates of
While ANOVA identified genes which show significant differences in expression profiles among the three TNBC subtypes, it could not identify differentially expressed sets of genes distinguishing the subtypes of TNBC. Therefore, we performed additional supervised analysis comparing gene expression profiles between individual subtypes of TNBC. We hypothesized that gene expression profiles significantly differ between individual subtypes of TNBC under study. Estimates of
Functional Relationship
To further refine the genetic susceptibility landscape and identify functionally related genes with similar patterns of expression profiles, we performed unsupervised analysis using hierarchical clustering on the set of genes which were significantly (
Figure 3 presents patterns of expression profiles of SNP-containing genes for each subtype and the controls. Figure 3A represents patterns of gene expression profiles for the 98 genes in normal-like subtype and controls. Figure 3B depicts patterns of gene expression profiles for the 101 genes in basal-like subtype and controls. Patterns of gene expression profiles for the 142 genes in basal subtype and controls are presented in Figure 3C. Patterns of gene expression profiles for the 159 genes showing consistent patterns across all the three subtypes are presented in Figure 4. In all four cases examined, genes were co-expressed and exhibited similar patterns of expression profiles. Interestingly, genes containing genetic variants with large effects and genetic variants replicated in multiple independent GWAS studies were co-expressed with genes containing genetic variants with small to moderate effects. Additional analysis using GO information revealed that genes containing SNPs associated with an increased risk of developing breast cancer are functionally related and are involved in the same biological processes and cellular components. A full catalogue of the physiological functions, biological processes, and cellular components in which the genes containing SNPs associated with risk for breast cancer are involved is presented in Table C (provided as supplementary data). These analyses confirmed our hypothesis that genes containing SNPs associated with an increased risk of developing breast cancer are functionally related. This is a significant finding given that traditional single-SNP GWAS analysis does not provide information about the functional relationship of genes containing SNPs associated with risk for developing breast cancer. Importantly, these data indicate that it is reasonable to use gene expression data as an intermediate phenotype to assess the association of GWAS information with TNBC and to gain biological insights about the broader context in which SNPs operate.

Patterns of gene expression profiles for genes containing SNPs associated with an increased risk associated with an increased risk of developing breast cancer, which were highly significantly (
To assess the significance of SNP-containing genes as potential biomarkers, we examined their functional relationship with high-penetrance genes (

Patterns of gene expression profiles for the 159 genes containing SNPs associated with an increased risk of developing breast cancer, which were highly significantly (
Association of Snp-Containing Genes with Novel Genes
One of the limitations of GWAS is that the susceptibility loci identified thus far are few and explain only a small fraction of the variation. This begs the question of where the missing variation is located. It is plausible that there are potentially many yet-to-be discovered common susceptibility alleles with smaller effects missed by GWAS. Even if the genes containing genetic variants associated with an increased risk of developing breast cancer are identified, it may not be obvious which genes mediate their biological effects. Therefore, key driver genes may be overlooked and important pathways may be missed by focusing solely on genes containing genetic variants associated with an increased risk of developing breast cancer. To address this question, we performed supervised analysis on the non-prioritized data set comparing gene expression profiles between each TNBC subtype and controls in order to identify novel genes which are significantly differentially expressed and co-expressed with SNP-containing genes. This analysis produced a set of 97 significantly (

Patterns and clusters of gene expression profiles for the 256 genes (159 SNP-containing genes and 97 novel genes not identified by GWAS, highly significantly associated with TNBC) in different subtypes of TNBC and controls.
Biological Pathways and Gene Networks
The second objective of this study was to investigate the broader context in which genetic variants and genes associated with TNBC operate and to identify gene regulatory networks and biological pathways enriched for SNPs, which are involved in TNBC. We hypothesized that genes containing SNPs associated with an increased risk of developing breast cancer, which are significantly associated with TNBC, interact with each other and with other genes not identified through GWAS. To test this hypothesis, we performed network analysis and pathway prediction using all 259 genes (SNP-containing and novel genes) as described in the methods section. We identified five top networks with the highest scores (predicted scores ranging from 28 to 51). The five top regulatory networks identified contained genes with multiple overlapping functions and involved multi-gene pathways. The first (network 1, score 51) contained genes involved in DNA repair, DNA mismatch repair, DNA replication and recombination, cell cycle, and nucleic acid metabolism. The second (network 2, score 41) produced genes involved in the DNA replication, recombination and repair, cellular compromise, and cell cycle. The third (network 3, score 33), contained genes involved in cell cycle, cellular development, cellular growth, and proliferation. The fourth (network 4, score 32) contained genes involved in cancer and organismal development. The fifth (network 5, score 28) contained genes involved in apoptosis. In all the networks identified, SNP-containing genes significantly associated with TNBC and other genes were functionally related.
To further refine the genetic susceptibility landscape, we mapped the genes onto the networks focusing on the most significant network. Figures 6–8 show the gene regulatory networks of SNP-containing genes and genes not identified through GWAS analysis. Network modeling and visualization revealed that SNP-containing genes significantly associated with TNBC interact with each other and with other genes not identified by GWAS confirming our hypothesis. Network analysis further revealed that genes containing SNPs with large effects and SNPs replicated in multiple independent studies interact with genes containing SNPs with small effects and not replicated. Among the identified SNP-containing genes were
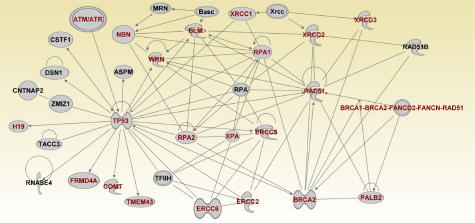
(Network 1). Gene regulatory networks containing multigene pathways involved in DNA replication, recombination, and repair, cell cycle, nucleic acid metabolism.

(Network 2). Gene regulatory network containing multigene pathways involved in DNA replication, recombination, and repair, cellular compromise, and cell cycle.

(Network 3). Gene regulatory network containing multigene pathways involved in cell cycle, cellular development, cellular growth and proliferation.
A close examination of the genes in the networks on the basis of molecular and cellular function revealed 66 genes which are involved in DNA repair, DNA mismatch repair, and base-excision repair to be the most significant (
To further determine the broader context in which genes containing genetic variants operate and to establish the functional bridges between GWAS findings and biological pathways relevant to TNBC, we performed pathway prediction. Specifically, we mapped the genes significantly associated with TNBC onto the pathways in the IPA database. The predicted pathways were ranked on predicted
Figure 9 shows the SNP-containing genes mapped to the

BRCA pathway showing the role of BRCA1 in DNA damage response and crosstalk with other biological pathways, notably the P53 pathway.
To further explore the role of SNP-containing genes in biological pathways relevant to TNBC, we explored the role of CHK proteins in the cell cycle checkpoint control pathway (Fig. 10). This pathway contained 6 SNP-containing genes

CHEK kinase pathway showing the role of CHEK proteins in cell cycle checkpoint control, and crosstalk with other pathways notably the P53 and BRCA pathways.
In all predicted pathways enriched for SNPs, genes containing SNPs with large effects and SNPs replicated in multiple studies were found to be interacting with genes containing SNPs with small to moderate effects. This is a significant finding given that overall, only a small number of statistically unimpeachable, common low-penetrance breast cancer susceptibility alleles have thus far been reported in TNBC in different populations.16,17 The identification of many multigene pathways enriched for SNPs indicates that many loci and pathway crosstalk may be involved in the pathogenesis of TNBC. The association of the DNA mismatch repair genes and pathways with TNBC is of particular interest both because ensuring fidelity of DNA replication is central to preserving genomic integrity and because DNA mismatch repair is critical for maintaining the fidelity of replication.
51
The functional relationship, similarity in patterns of gene expression profiles, and interactions in gene regulatory networks and pathways between different sets of genes, highlights the complexity of the molecular mechanisms involved in TNBC. An exciting result with potential therapeutic and clinical significance in this study is that genes with high penetrance (
Discussion
The primary goal of this study was to determine whether genes containing SNPs associated with an increased risk of developing breast cancer are associated with and could stratify different subtypes of TNBC. In addition, we sought to identify molecular pathways and networks relevant to TNBC. Our analysis establishes the association between genes containing SNPs associated with an increased risk of developing breast cancer and TNBC. It further identifies gene regulatory networks and biological pathways enriched for SNPs, which are involved in TNBC. This is a significant finding because to date, very few genetic variants and genes associated with an increased risk of developing TNBC have been reported.16,17 Many examples highlight the power of transcription profiling (signatures) in informing an understanding of the molecular basis and stratify subtypes of TNBC,8,13,14 as well as in the prediction of clinical outcomes.8,14,15,52,53 However, although these studies have made great strides in deciphering the molecular basis of TNBC, they have been unsuccessful in determining which genes have causative roles as opposed to being consequences of breast cancer state. 2 Recently our group reported integration of GWAS with gene expression data.2,54,55 However, this is the first study to link genes containing SNPs associated with an increased risk of developing breast cancer both with the TNBC intermediate phenotype and with the identification of biological pathways enriched for SNPs, which are involved in TNBC.
Importantly, this study indicates that combining GWAS information with gene expression data provides a powerful approach to identification of potential predictive biomarkers involved in TNBC. Predictive markers in TNBC will be particularly important because in the absence of effective therapy, these tumor subtypes tend to have poor prognosis. 8 Although, the ability to interpret the direct effects of the genetic variants on genes and pathways remains a challenge, this does not diminish the power of integrative analysis presented here to provide insights about the broader context in which genetic variants operate. It also establishes functional bridges between GWAS findings and the TNBC intermediate phenotype. Another novel feature of this integrative genomics approach is that it allows identification of additional genes which could not be identified using GWAS alone.
The practical significance of our approach is that it can be used to identify candidate genes to prioritize for sequencing. By prioritizing and evaluating SNP-containing genes using gene expression profiles, we were able to identify not only the most promising genes but also candidate pathways. This study therefore has the important goal of maximizing the yield and biological relevance of further downstream screens, experimental validation, functional studies, and targeted sequencing (by focusing on the most promising genes and biological pathways). Although we did not perform experimental validation, we used functional and co-expression analysis along with pathways prediction and network modeling in order to prioritize the genes on the basis of putative links to other genes–-notably high, moderate, and low penetrance genes that have more established roles as key drivers of breast cancer.
1
Future research directions for prioritization will focus primarily on broadening the scope of this study beyond transcription profiling of SNP-containing genes to include sequence information. A key opportunity and component will be the prioritization of genetic variants and associated genes for the purposes of next generation sequencing and elucidating the impact of genetic variants on gene and pathway function. Such work was beyond the scope of this report, but it is ongoing and will be reported elsewhere. Although we did not perform sequencing, some of the genes identified in this study (notably
One caveat is important in this study. We observed significant heterogeneity in patterns of expression profiles and overlap was incomplete. This suggests to the research community that to make headway against TNBC, we researchers must first come to grips with the burgeoning data from this and other studies, 8 showing that this subgroup of breast cancer is highly heterogeneous. The heterogeneity observed in this study indicates that TNBC is not a single disease entity and that genetic susceptibility could potentially be TNBC subtype-specific. Thus, identification of predictive risk markers must be conducted with that in mind.
It is worth noting that until recently, the only candidates for defining TNBC were mutations in
The results found in this study provide convincing evidence that genes containing SNPs associated with an increased risk of developing breast cancer are associated with TNBC and the identification of biological pathways enriched for SNPs, which are involved in TNBC. However, several limitations of this study must be acknowledged. First, we used publicly available GWAS information and gene expression data. The results could potentially be influenced by factors contained in use of such information and are beyond the scope of this report. Second, we did not investigate allele-specific gene expression. Current knowledge about how SNPs identified by GWAS–-particularly those mapped to noncoding and inter-genic regions–-regulate gene expression and pathways remains sketchy at best. However, we can now at least begin to understand the broader context in which they operate. Moreover, because the disease-causing alleles are likely uncommon, it is unlikely that they will be identified by association studies.
1
Integrative genomics provides a powerful approach for identifying candidate genes for further downstream screening. Although we did not investigate allele-specific expression, an earlier report on breast cancer confirmed that alleles in genes
Importantly, the majority of the GWAS information, as well as gene expression data used in this study, were derived from Caucasian populations. TNBC preferentially affects young African-American women. 60 While TNBC represents about 15% to 20% of all diagnosed breast cancers in the general US population, it constitutes about 30% in the African-Americans. 61 In fact, a recent study revealed populations differences and over-representation of TNBC in indigenous African women. 62 It is conceivable that genetic variants may confer population-specific risks depending on exposure. In the absence of gene expression data on the African-American population, we were not able to address that question, though it warrants investigation in future. Accordingly, we view this study as exploratory and the results found here cannot be generalized to different ethnic populations.
In conclusion, this study provides convincing evidence that genes containing SNPs associated with an increased risk of developing breast cancer are significantly associated with and could stratify the TNBC intermediate phenotypes. The study further reveals molecular pathways and networks enriched for SNPs, which are involved in TNBC. Based on these results we recommend that an integrative genomics approach combining GWAS information and gene expression data provides a unified and powerful approach to identification of potential biomarkers and molecular pathways in TNBC. More studies directed at understanding how the genetic variants regulate gene expression in target populations, notably the African-American population, are needed.
Author Contributions
Conceived and designed the experiments: CH. Analysed the data: CH, RK, KB. Wrote the first draft of the manuscript: CH. Contributed to the writing of the manuscript: CH, AP, AB, JM, KB, LM. Agree with manuscript results and conclusions: CH, RK, KB, AP, AB, JM, LM. Jointly developed the structure and arguments for the paper: CH, RK, LM. Made critical revisions and approved final version: CH, AB, LM. All authors reviewed and approved of the final manuscript.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgements
This study was supported by funding from the University of Mississippi Cancer Institute, to whom the authors are very thankful.