
Abstract
Lung cancer has a high mortality rate in men and women worldwide. Approximately 15% of diagnosed patients with this type of cancer do not exceed the 5-year survival rate. Unfortunately, diagnosis is established in advanced stages, where other tissues or organs can be affected. In recent years, lineage-specific transcription factors have been associated with a variety of cancers. One such transcription factor possibly regulating cancer is RUNX2, the master gene of early and late osteogenesis. In thyroid and prostate cancer, it has been reported that RUNX2 regulates expression of genes important in tumor cell migration and invasion. In this study, we report on RUNX2/p57 overexpression in 16 patients with primary non-small cell lung cancer and/or metastatic lung cancer associated with H3K27Ac at P1 gene promoter region. In some patients, H3K4Me3 enrichment was also detected, in addition to WDR5, MLL2, MLL4, and UTX enzyme recruitment, members of the COMPASS-LIKE complex. Moreover, transforming growth factor-β induced RUNX2/p57 overexpression and specific RUNX2 knockdown supported a role for RUNX2 in epithelial mesenchymal transition, which was demonstrated through loss of function assays in adenocarcinoma A549 lung cancer cell line. Furthermore, RUNX2 increased expression of epithelial mesenchymal transition genes VIMENTIN, TWIST1, and SNAIL1, which reflected increased migratory capacity in lung adenocarcinoma cells.
Introduction
Lung cancer is the major cause of cancer death in the world, resulting in 1.69 millions of deaths per year.1,2 Lung cancer is histopathologically classified into two great groups: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC), where this later one is the most frequent with 80%–85% patients presenting this type of cancer. In addition, NSCLC is subdivided into three subgroups: large cell carcinoma, squamous cell carcinoma, and the most frequent type adenocarcinoma, with 38% reported cases.3–5
In cancer development and progression, normal cells suffer a series of molecular and phenotypic changes, granting them tumor cell phenotype. As described by Hanahan and Weinberg, 6 multistep development of human tumors includes six features: sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, induced angiogenesis, capability of eliciting replicative immortality, and activation of invasion and metastasis.
Epithelial mesenchymal transition (EMT) is an event where cells lose their epithelial properties, while acquiring mesenchymal characteristics.7–9 During this process, it has been demonstrated that there are strong changes in epithelial and mesenchymal gene expression, regulated by important transcription and signaling factors. 9 In general, EMT is characterized by low E-CADHERIN and increased N-CADHERIN and VIMENTIN expression. For different types of cancer, EMT has been associated with a poor patient outcome and resistance to treatment.8,10 E-CADHERIN (CDH1) and N-CADHERIN are molecules of the CADHERIN family, fundamental in adherence and tissue maitenance. 10 In addition, several transcription factors (TFs), such as TWIST1 and SNAIL1, have been reported to participate in E-CADHERIN suppression, directly binding to their promoter region, thus actively participating in cell migration and invasion.11–14
In past years, lineage-specific TFs have been reported to take part of pathophysiological changes, favoring tumor cell proliferation and invasion. 15 RUNX TFs are key regulators of lineage specification. They are associated with major developmental pathways, such as transforming growth factor-β (TGF-β), WNT, Indian hedgehog, NOTCH, receptor tyrosine kinases, and mammalian STE20-like protein kinase (MST)–yes-associated protein 1 (YAP1). 16 RUNX proteins have multiple roles in cancer pathogenesis: in some cancers, they are strong tumor suppressors, while in others, they are oncogenic. Uncontrolled RUNX expression is frequently observed in diverse cancer types and has been shown to play major roles in the carcinogenic process. 16
During early and late embryogenesis, RUNX2 is essential for osteoblast cell differentiation and bone development. 17 In humans, RUNX2 encodes two isoforms from two alternative promoters, namely RUNX2-type I (also called Cbfα1/p56) and RUNX2-type II (also called Cbfα1/p57). 18 RUNX2/p56 is controlled by the proximal P2 promoter and represents the major RUNX2 isoform in tumor cells. 17 In contrast, RUNX2/p57 is controlled by the distal P1 promoter. There is a differential distribution in type I and II isoforms, not only regarding their spatio-temporal expression, but also with respect to the tissues that express them. In addition, expression can vary depending on species. Observations at tissue level revealed both isoforms exist in bone and lung, whereas RUNX2/p56 is widely expressed in heart, brain, spleen, and skeletal muscles. 18 RUNX2 expression in breast, colon, and thyroid cancer has been demonstrated to be important in tumor cell invasion.19–21 In prostate cancer, Yuen et al. 22 demonstrated RUNX2 was correlated with increased prostate-specific antigen (PSA). In lung cancer, Tandon et al. 23 demonstrated RUNX2 overexpression was associated with bone morphogenetic protein-3B (BMP-3B/GDF10) silencing. However, it is unknown which RUNX2 isoform expression is altered and what is the contribution to EMT in lung cancer.
In this study, we detected increased in RUNX2/p57 expression in patients with primary and metastatic NSCLC. Interestingly, results demonstrated increases in mRNA expression were greater in patients with primary lung cancer in comparison with patients with metastatic cancer. For all analyzed patients, transcriptional activation of RUNX2/p57 was associated with enrichment of H3K27Ac at P1 gene promotor region. However, it is important to highlight that in four analyzed patients, additional enrichment of H3K4Me3 occurred simultaneously with enzymes of the COMPASS-LIKE complex: WDR5, MLLl2, MLL4, UTX. On the other hand, silencing experiments allowed to establish RUNX2 TF participated in transcriptional regulation of E-CADHERIN, VIMENTIN, TWIST1, and SNAIL1, affecting A549 lung adenocarcinoma cell migratory capacity.
Materials and methods
This study included tissue specimens from 16 patients (10 primary lung cancer and 6 secondary lung cancer or metastatic cancer) with histopathologically verified lung masses (Table 1). Patients underwent surgical resection between January and May 2017 at the Hospital Universitario San Ignacio/Bogotá, Colombia. Research was performed under the Colombian Ministry of Health guidelines (No. 008430, 1993) and approved by the Pontificia Universidad Javeriana School of Medicine Ethics Committee. All procedures were carried out after written signed informed consent was obtained from all participants.
Clinical characteristics of all cancer patients.
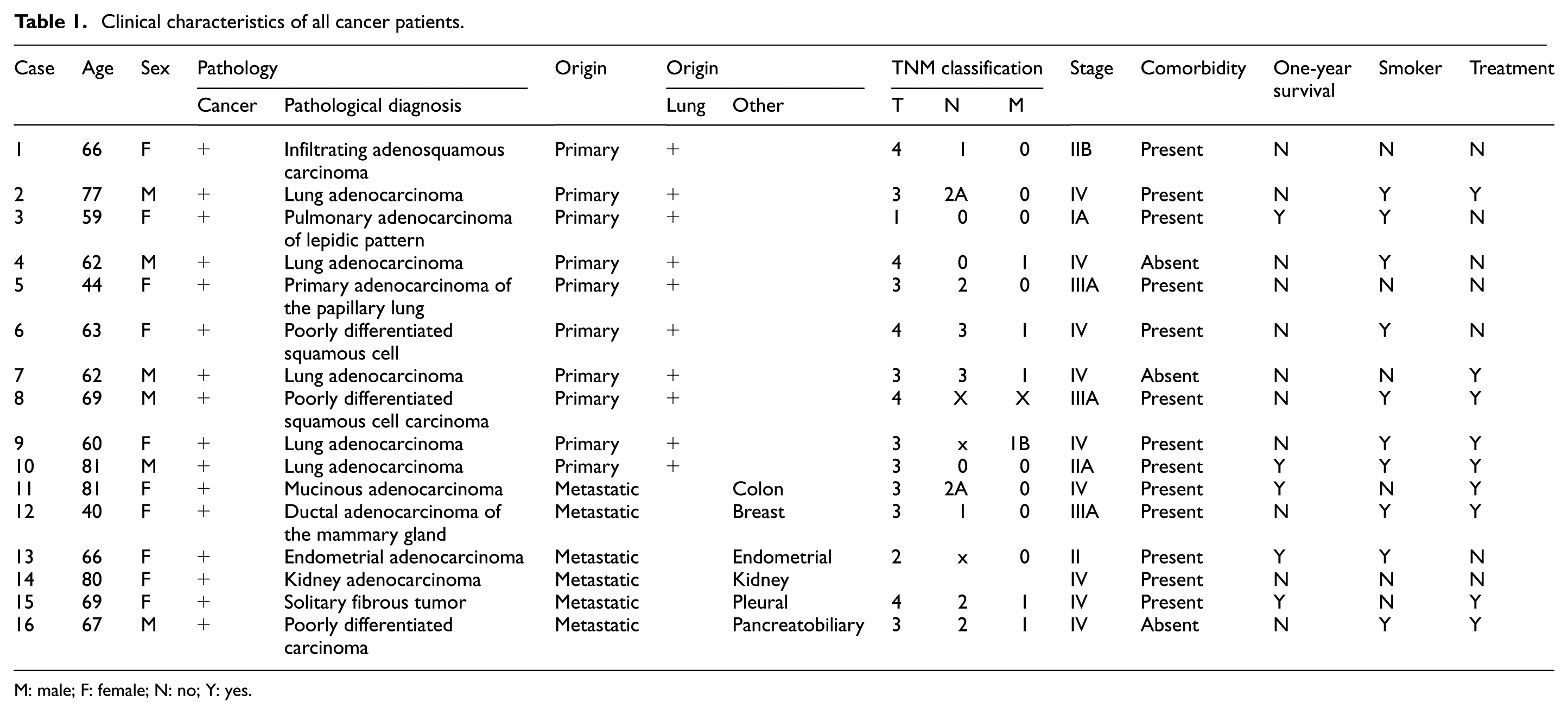
M: male; F: female; N: no; Y: yes.
Cell culture
Primary cell culture was established from biopsies following our protocol from previous research. 24 From these samples, gene expression and chromatin immunoprecipitation (ChIP) analyses were performed.
Human lung adenocarcinoma cell line A549 (ATCC®; Manassas, VA) was cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS) and 5% antibiotics (ampicillin and streptomycin) and incubated at 37°C in humidified 5% CO2 atmosphere. Cells were grown to 80% confluence to perform silencing and invasion assays. To induce the EMT process, A549 cells were treated with TGF-β1 (10 ng/mL, Abcam 50036) according to Zhang et al. 25
Lentivirus production and infection of A549 cells
HEK293FT cells (Life Technologies) were grown in 60 mm culture plates to reach 80% to 90% confluence. To transfect cells, Lipofectamine 2000 (Life Technologies) was used with pCMV-VSVg, pCMVdR8.91, and pLKO.1-shRNA plasmids following manufacturer’s instructions at a ratio of 1:2:3, respectively, with maximum total DNA of 10 µg per plate. pLKO.1 EV (sh-Ctrl) was used as control. Plasmids were donated by Dr Martín Montecino, Universidad Andrés Bello, Santiago, Chile. After 16–18 h, culture medium was replaced, and cells were maintained at 32°C for 48 h. Supernatants containing pseudo-typed particles were collected and filtered through a polyvinylidene fluoride filter (0.45 µm pore size). Supernatant aliquots were immediately stored at 80°C. 17 A549 cells were plated in 6-well culture plates and infected for 48 or 72 h with shRUNX2 or pLKO.1 EV (empty vector) with 5.779 × 109 copies/mL viral particles and 0.005 mg polybrene.
Nuclear extracts and protein expression analyses
Nuclear extracts were prepared as previously reported. 24 Total protein was quantified by the Bradford assay using bovine serum albumin as standard. For Western blot assays, 15 µg total protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis transferred to nitrocellulose membranes and immunoblotted. Primary antibodies used were RUNX2 (NBP2-24755SS; Novus Biologicals) and TFIIB C-18 (sc-225; Santa Cruz Biotechnology) as loading control. Immunoblots were visualized by enhanced chemiluminescence system (PerkinElmer Life Sciences). Densitometry was performed on scanned immunoblot images using ImageJ gel analysis tool. 26
Reverse transcriptase and quantitative real-time polymerase chain reaction
Total RNA was extracted with TRIzol (Life Technologies) according to manufacturer’s protocol. An equal amount of each sample (2 µg) was used for reverse transcription. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using SYBR Green I Master real-time PCR kit (Roche). Data are presented as relative mRNA levels of the gene of interest normalized to GAPDH mRNA levels.
ChIP
To identify regulatory components mediating epigenetic changes related with RUNX2 transcriptional control during lung cancer progression, ChIP assays were performed in primary cell culture. To this end, cross-linked chromatin samples were employed as described in Rojas et al. 27
Re-ChIP assays were performed as previously described. 28 Briefly, immunoprecipitated complexes obtained by ChIP were eluted by incubation for 30 min at 37°C in 25 μL of 10 mM dithiothreitol. After centrifugation, supernatant was diluted 20 times with sonication buffer and subjected to ChIP procedure with the second antibody.
Invasion assay
A549 cells infected with shRUNX2 or shCtr were cultured separately until reaching 80% confluence. Cells were then washed three times with phosphate-buffered saline (PBS) and cultured in serum-free media overnight before being subjected to an in vitro extracellular matrix (ECM) protein invasion assay. Invasion assay was conducted using BioCoat Matrigel Invasion Chambers with 8 μm pores (BD Biosciences, Bedford, MA) according to manufacturer’s instructions. Briefly, 70,000 cells were resuspended in fresh serum-free media and seeded into the upper chamber of a 24-well transwell plate, while the lower chamber contained fresh culture media with 5% FBS with or without 10 ng/mL TGF-β1. Cells were allowed to invade for 48 h (37°C, 5% CO2 humidified atmosphere). Chambers were then washed with PBS. Those cells that did not invade through the membrane were removed. Cells that invaded to the lower surface of the membrane were fixed with cold methanol, stained with 0.2% crystal violet, and examined. Cells on each membrane were counted in not less than five fields under a light microscope.
Statistical analyses
To compare significant changes with respect to control (noncancerous tissue (NCT)) for ChIP assays, one-way analysis of variance analysis followed by Dunnett’s post hoc test was performed to determine differences. To establish gene expression differences, unpaired Student’s t test was carried out. In all figures, error bars represent the standard error of the mean (*p < 0.05, **p < 0.01, ***p < 0.001).
Exploratory analyses were performed to determine whether RUNX2/p57 expression levels were associated with cancer stage, 1-year survival, smoking history, and patient age. Using the median value distribution of RUNX2/p57expression levels, participants were grouped into high RUNX2/p57 and low RUNX2/p57 levels; clinical parameters were compared between groups. To establish statistically significant differences between groups, a Student’s t-paired test was used. Data bivariate normality distribution was then determined, and according to the type of variables, a polychoric correlation, polyserial correlation, or Pearson correlation between variables was performed. The null hypothesis in all cases was rejected with a level of significance of p < 0.03. Statistical analyses were carried out employing R statistical software®.
Results
RUNX2/p57 overexpression in tissues of patients with primary lung cancer and secondary cancer with lung metastasis
Real-time qRT-PCR was performed to quantify RUNX2/p56 and RUNX2/p57 isoform expression in cancer tissues obtained from 16 cancer patients, 10 with primary lung cancer (patients 1–10) and 6 with secondary cancer with lung metastasis (patients 11–16). We used two samples obtained from non-tumor lung tissue (NCT) as controls. For all samples, GAPDH and β-actin were used as internal controls to normalize differences in total RNA levels for each sample. Results revealed that 87% (14 out of 16 patients) of tumor tissues had higher RUNX2/p57 expression levels in comparison with non-tumor tissue (Figure 1 and Supplemental Figure 1). Similarly, expression of RUNX2/p56 in tumor tissue showed a significant increase in 3 patients. However, it is important to note that overexpression levels were below those detected for RUNX22/p57 isoform (Figure 1(a) and (b); Supplemental Figure 1(A) and (B)). Figure 1(c) and (d) illustrates protein expression levels by Western blot for seven patients and a non-tumor sample. Results revealed RUNX2/p57 isoform expression levels were greater in comparison with RUNX2/p56. Figure 1(e) depicts percentage increase in RUNX2/p57 in comparison with RUNX2/p56 isoform.
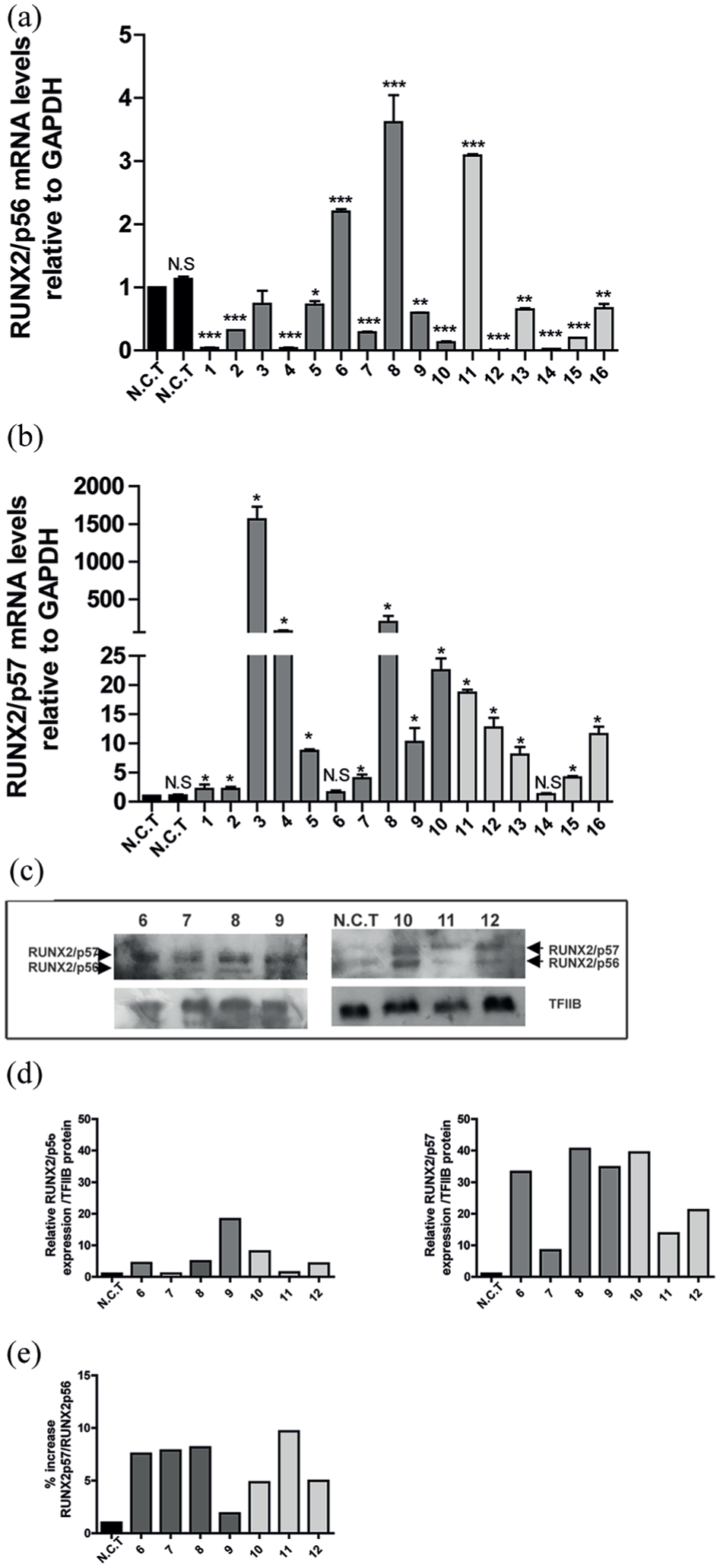
RUNX2/p57 overexpression in tissues of patients with primary lung cancer and secondary cancer with lung metastasis. RUNX2/p56 (a) and RUNX2/p57 (b) relative mRNA expression in lung cancer tissues of patients with primary lung tumor (patients 1–10) and secondary tumors (patients 11–16) compared with noncancerous tissue (NCT). mRNA levels were quantified by qRT-PCR and normalized to GAPDH mRNA. Statistical analyses were performed with respect to NCT. *p < 0.05; **p < 0.01; ***p < 0.001. (c) RUNX2/p56 and RUNX2/p57 protein isoform expression. TFIIB protein levels were used as loading control. RUNX2/p56 and RUNX2/p56 Western blot densitometry was performed using ImageJ. (d) RUNX2-p57/RUNX2-p56 protein percentage increase. (e) Percentage increase in RUNX2/p57 in relation to RUNX2/p56.
Correlation between clinical parameters and expression levels of RUNX2/p57
Table 1 describes study subjects with their clinical characteristics. Data on cancer stage, smoking history, and age were collected at the time of enrolment and after 1 year of patient survival. Using RUNX2/p57 expression level median value distribution, which was 9.47, participants were grouped in high and low RUNX2/p57 levels and clinical parameters for comparison between groups. No significant differences were observed for comparisons between participant age, smoking history, and cancer stage and 1-year survival.
Participants were grouped based on cancer origin, with 10 (n = 10) participants with primary lung cancer and 6 (n = 6) participants with metastatic lung tumors. Using RUNX2/p57 expression median value distribution for each group, participants were grouped into high RUNX2/p57 and low RUNX2/p57 levels and clinical parameters were compared between groups. For the lung cancer metastatic group, no differences were observed for comparisons between participant age, smoking history, and cancer stage and 1-year survival.
For the primary lung cancer group, no differences were observed in comparisons between participant age, smoking history, cancer stage, and 1-year survival. Interestingly, a significant difference was observed between high and low RUNX2/p57 levels for participants with cancer stages II and IV under 16.4 RUNX2/p57 expression level median value distribution and participants with stage III above RUNX2/p57 expression level median value distribution. Due to the number of participants, Student’s t-paired test statistical analysis between groups could not be performed. However, correlations between RUNX2/p57 expression levels and age, smoking history, and cancer stage and 1-year survival and primary lung cancer were performed. No correlations between age, smoking history, and 1-year survival and RUNX2/p57 levels were observed. A strong tendency associated with high RUNX2/p57 expression in stage III was noted (Figure 2). Considering the very small sample size, we believe these findings should be analyzed with caution.

Correlation between expression of RUNX2 and lung cancer stage. There was a strong increased RUNX2 expression tendency in participants at clinical stage III and low RUNX2 expression in participants at clinical stages II and IV. Black boxes represent 50% of data around the median (white circle). Purple areas show data value frequency around the median.
RUNX2/p57 expression in lung tumor cells involves changes in epigenetic histone marks
Transcription of RUNX2/p57 gene, a master regulator of osteoblast differentiation, is controlled by the P1 promoter sequence, specifically the 500-pb region most proximal to transcription initiation site, which suffers chromatin remodeling in mouse osteoblastic cells during RUNX2 expression. 27 This promoter region is highly conserved and has been shown to include regulatory elements that are functional within human, rat, and mouse osteoblastic cells. 27 To shed light on the cellular process controlling RUNX2/p57 expression in human lung cancer, contribution of epigenetic mechanisms was assessed. Tumor cells exhibited at the P1 promoter post-translational modifications at histone H3. Thus, enrichment at H3K27Ac was observed with H3K27Me3 reduced levels at the RUNX2 P1 promoter site (Figure 3(b) and (c)). Surprisingly, we found enrichment of H3K4Me3 in patients 8,9,12, and 13 (patients 8, 9, 12, and 13) (Figure 3(a)). Hence, this epigenetic mark corresponds to the activacion of the P1 promoter during tumor progression.

RUNX2 epigenetic transcriptional control. Histone post-translational modifications at RUNX2 P1 promoter region in lung cancer cells and secondary cancer cells with lung metastasis. Primary lung cancer cells of four patients (patients 7, 8, 9, and 10) and secondary cancer cells (patients 12 and 13) were used to perform ChIP assays. Antibodies against (a) H3K4Me3, (b) H3K27Ac, and (c) H3K27Me3 were used. Results are expressed as % input ± SEM using normal IgG as specificity control. Statistical analyses were performed with respect to NCT cells.
RUNX2 P1 promoter was recognized by epigenetic regulators during lung tumor progression
In order to identify regulatory components mediating epigenetic changes related with RUNX2/p57 transcriptional control during lung cancer progression, ChIP analyses were performed. WDR5, MLL2, MLL4, and UTX exhibited a significantly higher enrichment at the proximal RUNX2 P1 promoter sequence for patients 8, 9, 12, and 13 (Figure 4(a)–(d), respectively).

RUNX2 P1 promoter was recognized by chromatin-modifying enzymes in lung cancer cells and secondary cancer cells with lung metastasis. Primary lung cancer cells of four patients (patients 7, 8, 9, and 10) and secondary cancer cells (patients 12 and 13) were used to perform ChIP assays. Antibodies against chromatin-modifying proteins (a) WDR5, (b) MLL2, (c) MLL4, and (d) UTX were used. Results and statistical analyses are shown as described in previous figure legend. Normal IgG was used as specificity control. *p < 0.05; **p < 0.01; ***p < 0.001. (e) Re-ChIP assay was performed using chromatin extracted from the tumor of patient 9. This assay demonstrated RUNX2 P1 promoter co-occupancy by MLL2, MLL4, and UTX. The first ChIP assay was performed with antibody against UTX, and the second ChIP (Re-ChIP) was carried out with antibodies against MLL2 and MLL4. Controls for the Re-ChIP assay were performed with anti-rabbit and anti-mouse IgG (IgG/IgG).
We have recently shown that during osteoblast commitment, a MLL/COMPASS-LIKE complex plays a relevant role during RUNX2 transcription. 17 Next, it was determined whether additional subunits (in addition to UTX) of these H3K4 methyltransferase complexes were bound to the RUNX2 P1 promoter (Figure 4(e)). It was found that MLL2 and MLL4 were enriched at this RUNX2 P1 promoter sequence (Figure 4(e)).
Increase in RUNX2/p57 expression in A549 cell line tumor progression
To determine whether cells initiated an EMT process, first EMT key markers E-CADHERIN, N-CADHERIN, and VIMENTIN were quantified in cells treated with TGF-β1 (Figure 5(a)–(c); Supplemental Figure 2). Initiation of EMT after stimulation, was observed based on low E-CADHERIN epithelial marker expression was detected. In addition, overexpression of N-CADHERIN and mesenchymal marker VIMENTIN were observed. To assess RUNX2/p56 and RUNX2/p57 expression during EMT, mRNA and protein levels were quantified. Results revealed an increased in RUNX2/p57 expression and protein levels after cells were treated with TGF-β1 (Figure 5(d)–(f); Supplemental Figure 2). Moreover, RUNX2/p56 expression in TGF-β1 treated and untreated cells remained constant (Figure 5(d)–(f); Supplemental Figure 2).

Overexpression of RUNX2 in A549 cell line induced with TGF-β1 to promote EMT process. Human lung adenocarcinoma A549 cells were pre-cultured with or without 10 ng/mL TGF-β1 for 48 h. (a)–(c) E-CADHERIN, N-CADHERIN, and VIMENTIN mRNA levels in A549 cells. (d)–(f) RUNX2/p56 and RUNX2/p57 mRNA and protein levels in A549 cells with or without TGF-β1 treatment. Statistical analyses were performed with respect to A549 cells without TGF-β1.
Absence of RUNX2 was associated with decreased EMT markers and TWIST1 and SNAIL1
To determine the role of RUNX2 in EMT process, we knocked down RUNX2 using shRNA in TGF-β1 stimulated or unstimulated cells. As shown in Figure 6(a) and (b), utilized shRUNX2 affected both mRNA expression isoforms and proteins (RUNX2/p56 and RUNX2/p57). Expression of E-CADHERIN, N-CADHERIN, VIMENTIN, TWIST1, and SNAIL1 was quantified by qRT-PCR (Figure 6(c)–(g); Supplemental Figure 3). Likewise, for this analysis, EMT was evaluated in sh-Ctrl (control) treated cells and stimulated or unstimulated with TGF-β1 with the previously mentioned markers (Figure 6(c)–(g)). A decrease in E-CADHERIN after shRUNX2 treatment in TGF-β1 unstimulated cells was detected (Figure 6(c)). Moreover, a decrease in VIMENTIN expression was also observed in TGF-β1 treated and untreated cells (Figure 6(e)). In contrast, when RUNX2 was silenced, N-CADHERIN did not show any variation (Figure 6(d)). In addition, cells infected with shRUNX2 evidenced significant changes in TWIST1 and SNAIL1 gene expression (Figure 6(f) and (g); Supplemental Figure 3).
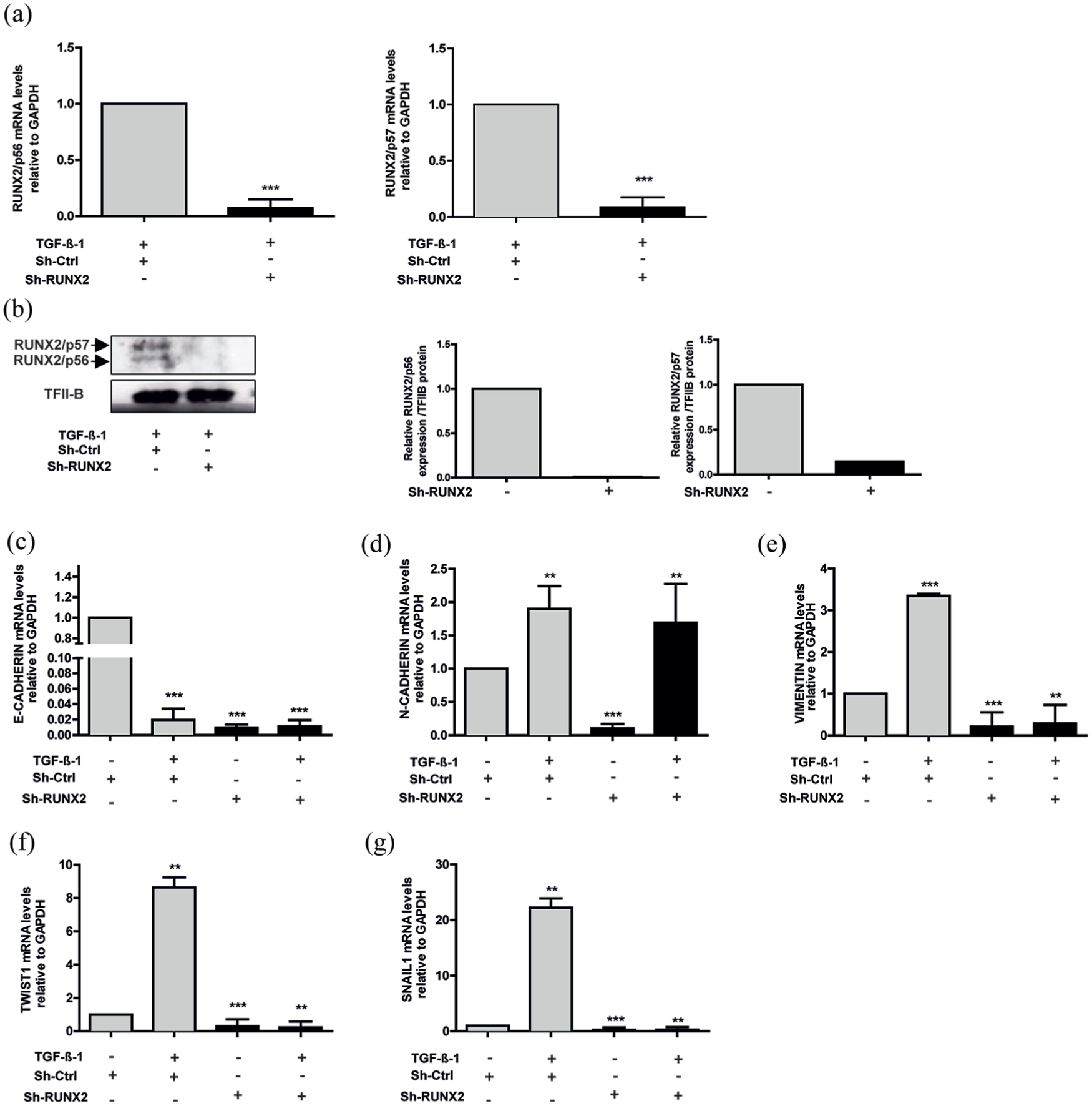
Knockdown of RUNX2 affects EMT marker expression. A549 cells were stimulated with or without TGF-β1 as described in Figure 4. A549 cells were infected with lentiviral particles coding for shRNAs against RUNX2. Effective down-regulation was confirmed by qRT-PCR (a) and Western blot (b) analyses 72 h post infection. TFIIB protein levels were used as loading control (c). E-CADHERIN, (d) N-CADHERIN, (e) VIMENTIN, (f) TWIST1, and (g) SNAIL1 mRNA levels were quantified by qRT-PCR 72 h after infection. Statistical analyses were performed with respect to cells infected with virus generated with the pLKO.1 empty vector (sh-Ctrl.).
Absence of RUNX2 affect A549 lung adenocarcinoma invasion capacity
To evaluate RUNX2 A549 lung adenocarcinoma cell capacity to invade, a transwell system was employed (Figure 7). Our results show that shRUNX2 completely inhibited TGF-β-induced invasion, suggesting RUNX2 may be responsible for increased invasion. Quantification of the assay is illustrated in Figure 7. For statistical analysis, control cells were transduced with sh-Ctrl and treated (+) or untreated (–) with TGF-β1.

Effect of RUNX2 knockdown on transwell migration and invasion assay. (a) A549 cells were stimulated with or without 10 ng/mL TGF-β1 and infected with lentiviral particles coding shRNAs against RUNX2. Cells penetrating the membrane were fixed and stained after 48 h as described in experimental procedures. Migrating cells were counted after additional incubation with 10 ng/mL TGF-β1 for 48 h.
Discussion
In general, cell identity is regulated by TFs that recognize specific sequences in the genome, and thus regulate gene expression.29,30 Almost 50% of the TFs encoded in the genome are expressed in any cell type; however, a smaller number of master TFs are sufficient to establish control of gene expression programs, also known as lineage regulators that define cell identity.29,30 In recent years, these master TFs have been described to participate in neoplastic processes. Activation of a master TF, normally expressed early in a specific lineage, can alter core regulatory circuitry and activate additional genes that are regularly expressed in more embryonic states.29,30
It has been described RUNX2, an osteogenic master TF, is engaged in tumor progress, through direct or indirect regulation of genes favoring migration and invasion capacity in different types of cancer.19,23 In the present study, significantly higher RUNX2/p57 expression was demonstrated in samples of primary or metastatic lung cancer compared with samples from non-tumor tissue. Our results demonstrated mRNA did not precisely correlate with protein levels. Regarding the above mentioned, it has been demonstrated that transcription and translation are far from having a linear and simple relationship. Different mechanisms involving cis-acting and trans-acting mechanisms generate a large repertoire of posibilities, capable of enhancing or repressing protein synthesis from mRNA molecules. Different events may uncouple transcription and translation continuously or under certain conditions. 31 In 2013, Li et al. 20 reported RUNX2 overexpression in patients with NSCLC lung cancer. However, their work did not analyze RUNX2/p56 and RUNX2/p57 isoforms. For other types of cancer, such as breast, prostate and thyroid RUNX2/p56 overexpression has been reported. 20 Specifically, in studies using thyroid and breast cancer cell lines, overexpression has been associated with the presence of H3K27Ac and H3K4Me3 activator marks at the P2 promoter. 17
In the present study, correlation analyses between clinical parameters and RUNX2/p57 expression levels allowed to establish an association between RUNX2/p57 expression level and tumor stage. A strong tendency was established between RUNX2/p57 high expression levels and stage III. Stage II lung cancer is capable of spreading from the lungs to the lymph nodes or to nearby structures and organs, such as the heart, trachea, and esophagus. In contrast, in stage III, lung cancer has already metastasized to other areas of the body, 32 suggesting RUNX2 could be involved during the first steps of invasion, when cancer spreads to the lymph nodes and nearby organs. However, considereing the reduced number of patient samples, we believe that that these findings should be analyzed with caution.
In addition, to determine whether RUNX2/p57 overexpression was related with gene’s epigenetic chromatin modifications in associated histones, ChIP assay was carried out. RUNX2/p57 expression was related with H3K27Ac active enhancer mark enrichment at P1 promoter. Interestingly, 66.6% of analyzed patients by ChIP assay also had a H3K4Me3 active mark enrichment at the promoter region. This enrichment occurred simultaneously with enzyme recruitment of the COMPASS-LIKE complex (WDR5, MLL2, MLL4, and UTX) to the promoter region. Co-presence of UTX, MLL2, and MLL4 complex was additionally confirmed by re-ChIP.
Results are in agreement with those reported by Rojas et al.27,28 in 2015, where it was demonstrated in mouse mesenchymal cells RUNX2/p57 transcriptional regulation was determined by existence of a specific pattern of covalent modifications in histone residues. Therefore, during osteoblast differentiation, when the gene was transcriptionally active, enrichment of H3K27Ac and H3K4Me3 occurred in parallel with the COMPASS-LIKE complex (WDR5, MLL, UTX) recruitment. In contrast, when cells differentiated into myoblast lineage, enrichment levels of H3K27Ac and H3K4Me3 active marks were significantly decreased, with concomitant increase in repressor mark, such as H3K27Me3, among others.27,28
Participation of COMPASS-SET1 and COMPASS-LIKE complexes in cancer has been widely demonstrated. Mixed lineage leukemia (MLL) gene was first discovered as an oncogenic fusion resulting from seemingly random translocations in patients with hematological malignancies. 33 COMPASS complex is capable of catalyzing mono-, di-, and trimethylation on histone H3K4.
In mammals, COMPASS complex is grouped into three categories: COMPASS-Set, Mll1/Mll2 COMPASS (orthologs of Trx in Drosophila), and Mll3/Mll4 COMPASS (orthologs of Trr in Drosophila). 33 To ensure proper transcriptional modulation, a growing body of evidence points to a model, where H3K4 methylation is divided among COMPASS family members. 28 In this way, it has been demonstrated that Mll1/Mll2 is necessary to maintain H3K4 trimethylation levels, whereas Mll3/Mll4 is responsible for H3K4 monomethylation enrichment. 28 These findings would explain presence of Mll2 and Mll4 at the P1 promoter region of RUNX2 gene in samples from patients with primary and metastatic lung cancer.
Parallel to RUNX’s role in EMT during development, in different tissues, these proteins have been implicated in aberrant cancer EMT activation. The best characterized of these phenomena is RUNX2 involvement in breast and prostate cancer metastases. 34 In prostate cancer, it has been demonstrated that RUNX2 positively regulates EMT through direct regulation of SMAD3, SNAIL2, and SOX9 genes. In thyroid cancer, it has also been reported an association between RUNX2 and aberrant EMT through regulation of target genes, such as SNAIL2, VEGF, TWIST1, and MMP2. 34
To determine RUNX2 role in EMT process in lung cancer, RUNX2 loss of function by shRNA in lung adenocarcinoma A549 cell line was performed. This cell line is widely used to simulate in vitro EMT process under TGF-β1 stimulus, where cells acquire mesenchymal phenotype and their migration capacity is increased. 25 It is important to acknowledge cells infected with shRUNX2 demonstrated decreased expression in both of RUNX2 isoforms (RUNX2/p56 and RUNX2/p57). Our findings demonstrated EMT did not occur in cells infected with shRUNX2, as shown by low invasion capacity in transwell assay and decreased expression of VIMENTIN, TWIST1, and SNAIL1. On the other hand, incubation of cells with TGF-β repressed the expression of E-CADHERIN in the presence or absence of RUNX2.
An important EMT hallmark is loss of cell-to-cell adhesion molecule expression. E-cadherin is a central component of cell–cell adhesion junctions and is required for formation of epithelial tissues in the embryo and to maintain epithelial homeostasis in the adult. 35 Loss of E-cadherin expression is consistently observed at EMT sites during development and cancer. This loss has been found to increase tumor cell invasiveness in vitro and contributes to the transition of adenoma to carcinoma in animal models. 35 Several important proteins during development that induce EMT have been shown to act as E-cadherin repressors or activators. For example, Slug (also known as SNAI2), a member of the Snail family of transcriptional repressors, is capable of repressing E-cadherin expression and thereby triggering EMT, 35 suggesting that it may act as an invasion inducer. It has been acknowledged that both SNAIL and its family member SLUG are capable of repressing E-cadherin in epithelial cells via E-box elements in the proximal E-CADHERIN promoter. 35 Functional assays in the present study demonstrated that TGF-β repressed E-CADHERIN in the presence or absence of RUNX2. Hence, it is possible that E-CADHERIN may be under the control of other factors. Nonetheless, additional analyses are required to confirm this finding. At present, it is known that this gene is regulated by various TFs. Recently, Li et al. 36 demonstrated RUNX2 TF binds directly to E-CADHERIN promoter and positively regulates its activation during epithelial differentiation of adipose-derived stem cells in mammals. N-CADHERIN expression increased under TGF-β stimulation and decreased with shRUNX2; however, when TGF-β was added in cells expressing shRUNX2, expression was still increased. This result suggests possibly under TGF-β stimulation, other TFs could have regulated promoter activation.
In addition, we demonstrated an effect on TWIST1 and SNAIL1 gene expression, which participate in tumor cell migration, suggesting RUNX2 exerts a positive regulatory effect on both genes. Moreover, RUNX2 expression is essential in TWIST1 and SNAIL1 activation. Regulation of these genes by RUNX2 has been reported in thyroid tumor cells and lung adenocarcinoma in very advanced cancer stages.21,23,37 Furthermore, RUNX2 capacity to bind to TWIST promoter in neuroblastoma tumor cells has been described. 38
Our results also demonstrate RUNX2 participated in VIMENTIN expression, a mesenchymal marker, since shRUNX2 generated an ~80% decrease in VIMENTIN expression. In breast cancer cells, it has been reported that RUNX2 increases VIMENTIN and SNAIL2 expression.39,40 Likewise, VIMENTIN is an osteogenic differentiation master TF target gene, as is RUNX229,30,41 In conclusion, RUNX2 overexpression in lung cancer was related with EMT process, through direct regulation of E-CADHERIN, VIMENTIN, TWIST1, and SNAIL1 expression.29,30 Therefore, it could become a new therapeutic target for patients with NSCLC.
Supplemental Material
TUB_Supplementary_Figures – Supplemental material for Role of RUNX2 transcription factor in epithelial mesenchymal transition in non-small cell lung cancer lung cancer: Epigenetic control of the RUNX2 P1 promoter
Supplemental material, TUB_Supplementary_Figures for Role of RUNX2 transcription factor in epithelial mesenchymal transition in non-small cell lung cancer lung cancer: Epigenetic control of the RUNX2 P1 promoter by Angélica María Herreño, Andrea Carolina Ramírez, Viviana Paola Chaparro, María José Fernandez, Alejandra Cañas, Carlos Fabian Morantes, Olga María Moreno, Ricardo Elias Brugés, Juan Andrés Mejía, Fernando José Bustos, Martín Montecino and Adriana P Rojas in Tumor Biology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Pontificia Universidad Javeriana (PUJ) (6276, PUJ 7189) and M.M. was supported by FONDAP (15090007) and CONICYT-REDES (150109). A.M.H. was supported by COLCIENCIAS grant no. 705.2015-Jóvenes Investigadores e innovadores 2015.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.