
Abstract
Epithelial–mesenchymal transition is a crucial event for metastasis and could be mediated by several pathways such as phosphoinositide 3-kinase/Akt, mitogen-activated protein kinases, as well as many epigenetic regulators. Special AT-rich sequence-binding protein 2 is an epigenetic regulator involved in epithelial–mesenchymal transition and osteoblastic differentiation. It has been reported that the crosstalk between several pathways is responsible for the regulation of epithelial–mesenchymal transition in cancer cells. However, crosstalks between p38 and Akt pathways involved in epithelial–mesenchymal transition are still unknown. We recently reported that there is a crosstalk between p38 and Akt pathways in non-small-cell lung carcinoma cells, and this crosstalk is associated with E-cadherin and special AT-rich sequence-binding protein 2 expressions. Therefore, we aimed to determine whether this crosstalk has a mediator role in the regulation of epithelial–mesenchymal transition in non-small-cell lung carcinoma. Our results showed that inhibition of p38 leads to the disruption of this crosstalk via decreased expression of phosphatase and tensin homolog (PTEN) and subsequently increased activation of Akt in non-small-cell lung carcinoma cells. Then, we found that p38 inhibition upregulated special AT-rich sequence-binding protein 2 expression and reversed epithelial–mesenchymal transition in non-small-cell lung carcinoma cells. Furthermore, special AT-rich sequence-binding protein 2 knockdown abolished the effect of p38 inhibition on epithelial–mesenchymal transition in non-small-cell lung carcinoma cells. In conclusion, our results strongly indicate that the crosstalk between p38 and Akt pathways can determine special AT-rich sequence-binding protein 2 expression and epithelial character of non-small-cell lung carcinoma cells, and special AT-rich sequence-binding protein 2 is a critical epigenetic regulator for epithelial–mesenchymal transition mediated by p38 pathway in non-small-cell lung carcinoma. Our findings will contribute to illuminate the molecular mechanisms of the epithelial–mesenchymal transition process that has a critical significance for lung cancer metastasis.
Introduction
Epithelial–mesenchymal transition (EMT) is a significant event for invasion and metastasis of cancer cells. In this process, epithelial-derived cancer cells display decreased epithelial properties such as cell–cell junctions and apical–basolateral polarity and gains mesenchymal cell phenotype with enhanced invasive properties. The hallmarks of EMT are downregulation of epithelial markers such as E-cadherin and upregulation of mesenchymal markers such as N-cadherin and vimentin.1,2 EMT can be regulated by several signaling pathways including SMADs, signal transducers and activators of transcription (STATs), phosphoinositide 3-kinase (PI3K)/Akt, and mitogen-activated protein (MAP) kinases.2,3
Mitogen-activated protein kinases (MAPKs) are expressed in all cell types and highly activated in cancer cells. This pathway has an ability to regulate a variety of physiological processes such as cell growth, metabolism, and cell death. To date, six distinct groups of MAPKs have been identified in mammals: the extracellular signal–regulated kinases p38 MAPKs p38α/β/γ/δ, extracellular signal–regulated protein kinase (ERK)1/2, ERK3/4, ERK5, ERK7/8, and the c-Jun N-terminal kinase (JNK) 1/2/3. 4 Especially, p38 MAPK pathway has been implicated in the distinct stages of metastasis.4,5 The effects of p38 MAPK pathway on metastasis can be mediated via its interaction with different pathways. Estrada et al. reported that the crosstalk between ERK and p38 pathways mediates migration and proliferation of melanoma cells. 6 Staples et al. 7 indicated that the crosstalk between the p38 MAPK and JNK pathways mediated by MAP Kinase Phosphatase-1 plays a critical role in determining the fate of cancer cells with exposed DNA damaging agents such as ultra violet (UV) radiation and cisplatin.
Roles of the crosstalk between p38 MAPK and PI3K/Akt pathways in various cellular events are well known. Zarubin and Han 8 indicated that the crosstalk between p38 and PI3K/Akt pathways has a regulatory role in the orchestration of myoblast differentiation. Furthermore, Shen et al. 9 demonstrated that the crosstalk between p38 MAPK and PI3K/Akt pathways mediates endothelial NO synthase (eNOS) activation in endothelial cells. The emerging evidence of the interaction between p38 and PI3K/Akt pathways plays a critical role in physiological processes in cells and let us to think that this crosstalk might implicate in several stages of carcinogenesis. In our previous study, we reported that SATB2, which is a transcription factor and binds to AT-rich DNA sequences in nuclear matrix attachment regions is a novel regulator in EMT and invasion in non-small-cell lung carcinoma (NSCLC) cells. Therefore, in this study, we aimed to examine whether the crosstalk between p38 and PI3K/Akt pathways could mediate EMT process by regulating SATB2 expression in NSCLC cells.
Materials and methods
Cell culture and reagents
A549 and H1650 NSCLC cell lines were used in this study. A549 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), H1650 cells were cultured in RPMI 1640, and DMEM and RPMI 1640 were supplemented with 10% fetal bovine serum (FBS), 100 mg/mL penicillin, 50 mg/mL streptomycin, and 1 mM glutamine at 37°C in 5% CO2. SB203580 (specific p38 MAPK inhibitor) and LY294002 (a specific inhibitor of PI3K/Akt pathway) were purchased from BioVision(Milpitas, CA, USA). Antibodies of E-cadherin, N-cadherin, vimentin, Snail, Zeb1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies of SATB2, Slug, and Twist were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
A549 cells were seeded at 2 × 103 cells per well in 96-well tissue culture plates and incubated for 24 h in culture medium with 10% serum. Then, the cells were treated with p38 inhibitor SB203580 (500 nM) or dimethyl sulfoxide (DMSO) for 24 h, and cell viability was measured using Vybrant® MTT Cell Proliferation Assay Kit according to the manufacturer’s instructions (Thermo Fischer Scientific, Waltham, MA, USA). Formazan formation was quantified spectrophotometrically at 560 nm wavelength using a microplate reader.
Transient transfection
Expression vectors for the continuous active Akt (CA-Akt) were used in this study. A549 cells were seeded at a density of 2 × 105 cells per well in six-well plates and were transfected with the CA-Akt or mock vectors using Lipofectamine 2000 (Thermo Fischer Scientific, Carlsbad, CA, USA) according to the manufacturer’s instructions; 24 h after transfection, the medium was removed and cells were incubated in serum-free medium for an additional 24 h. Cell lysate preparation and western blot analysis were performed as described below.
Small interfering RNA transfection
A549 cells were transfected with SATB2 small interfering RNA (siRNA; 20 nM) or control siRNA (20 nM) using Lipofactamine2000 transfection reagent according to the manufacturer’s instructions; 24 h after transfection, cells were incubated in culture medium with 10% serum for an additional 24 h. Then, cell lysate preparation and western blot analysis were performed as described below.
Western blotting
The cell lysates were prepared in ice-cold radioimmunoprecipitation assay (RIPA) lysis solution. Western blot analysis was performed as previously described. 10
Wound-healing assay
The effects of inhibition of p38 on migration capability of A549 cells were assessed using a wound-healing assay. The cells were grown to 90% confluence on the six-well plate, washed with phosphate-buffered saline (PBS), and cultured for an additional 24 h with serum-free medium. The confluent cell monolayers were manually wounded by scraping the cells with a 20 μL pipette tip. To remove cell debris, the cells were washed with serum-free medium three times. Then, the cells were cultured for 24 h in serum-free medium with DMSO or p38 inhibitor SB203580 (500 nM). Photos were taken at 0, 16, and 24 h.
Cell invasion assay
The invasiveness of cells was evaluated by a Matrigel invasion chambers (BD Biosciences, San Jose, CA, USA) with 8-mm membrane pores. The media was removed and the cells were trypsinized and counted by hemocytometer and Trypan blue staining; 1.25 × 105 cells were seeded on upper chamber in serum-free media and treated with SB2035802 (500 nM) or DMSO for 24 h. In the lower chamber, 10% serum-added media plus SB2035802 (500 nM) was used as a chemoattractant. The cells were allowed to migrate for 24 h at 37°C. After incubation for 24 h, non-invaded cells on the upper side of the Transwell were removed with cotton swabs, and the invasive cells on bottom side of the Transwell were fixed with 100%methanol. Then, they were stained with 0.1% crystal violet solution and were counted under a light microscope in five random microscopic fields (magnification, 20×).
Statistical analysis
A statistical analysis was performed using a SPSS 17.0 version. Paired t-test was used to determine significance of the difference among the comparisons and p values less than 0.05 were considered statistically significant.
Results
The crosstalk between p38 and Akt signaling pathways might determine epithelial characters and SATB2 expression of NSCLC cells
It has been shown that the crosstalk between p38 and Akt signaling pathways implicates in several cellular processes in endothelial cells, macrophages, and myeloid cells, but there is no report showing the relationship between this crosstalk and induction of EMT for NSCLC cells. Therefore, we first examined the activation levels of p38 and Akt in A549 and H1650 cells in normal medium conditions. As shown in Figure 1(a), the level of active p38 is significantly higher in A549 cells than H1650 cells; However, activation level of Akt is lower in A549 cells compared to H1650 cells, indicating that there is a crosstalk between p38 and Akt pathways in NSCLC cells. Then, we wanted to compare the expressions levels of E-cadherin and SATB2 in A549 and H1650 cells by western blot analysis. Interestingly, we found that expression levels of SATB2 and E-cadherin are associated with p38-Akt crosstalk in A549 and H1650 cells, suggesting that the crosstalk between p38 and Akt signaling pathways might determine epithelial characters and SATB2 expression of NSCLC cells (Figure 1(b)).

The crosstalk between p38 and Akt signaling pathways might determine epithelial characters and SATB2 expression of NSCLC cells. (a) A549 and H1650 cells are cultured with normal growth medium containing 10% FBS, and then, cellular lysates were prepared in RIPA buffer and equal amount of proteins was fractionated using 10% polyacrylamide gel; blots were labeled with p-p38-, p38-, p-Akt-, and Akt-specific antibodies. The band intensities on films were quantified by densitometric analysis using Image Studio Lite Ver 5.2, and values of bands were normalized against p38 or Akt. To calculate fold inductions, the expression values of bands were divided by that of controls. (b) The same cellular lysates were fractionated using 10% polyacrylamide gel, and blots were labeled with E-cadherin-, SATB2- and GAPDH-specific antibodies. The band intensities on films were quantified by densitometric analysis using Image Studio Lite Ver 5.2, and values of bands were normalized against GAPDH. To calculate fold inductions, the expression values of bands were divided by those of controls.
Inhibition of p38 leads to increased Akt activation and upregulation of SATB2 expression
Because of the results that we mentioned above, we hypothesized that elevated activation of p38 could leads to decrease in activation of Akt and SATB2 expression in NSCLC cells. Therefore, we inhibited p38 activation by SB203580 (500 nM) in A549 cells which have high levels of activated p38. As expected, we observed that SB203580 treatment in A549 cells resulted in a decrease in p-p38 (phosphorylated-p38) level and an increase in p-Akt (phosphorylated-Akt) level (Figure 2). Thereafter, we wanted to explain why inhibition of p38 increases level of p-Akt. Thus, we examined the effect of p38 inhibition on expression level of phosphatase and tensin homolog (PTEN), which is a negative regulator for Akt activation. As shown in Figure 2, p38 inhibition strongly downregulated PTEN expression in A549 cells. To determine the effect of p38 inhibition on SATB2 expression, we evaluated the protein level of SATB2 in A549 cells treated with SB203580, and we found that p38 inhibition in A549 cells leads to upregulation of SATB2 expression (Figure 2). These results indicated that p38 inhibition can induce activation of p-Akt and SATB2 expression by downregulating PTEN expression. Then, we hypothesized that only increased activation of Akt might induce SATB2 expression in A549 cells. Therefore, we used A549 cells transfected with CA-Akt or mock vector. After 24 h from transfection, we evaluated the protein levels of p-Akt, Akt, SATB2, and GAPDH in A549 cells by western blot analysis. As expected, increased activation of Akt strongly induced SATB2 expression in A549 cells, indicating that there is a positive correlation between the level of p-Akt and SATB2 expression in NSCLC cells. Furthermore, we treated A549 cells with LY294002 that can make PI3K/Akt signaling pathway inactive. Inactivation of PI3K/Akt signaling pathway leads to downregulation of SATB2 expression, indicating that SATB2 expression is regulated by PI3K/Akt pathway in NSCLC cells.
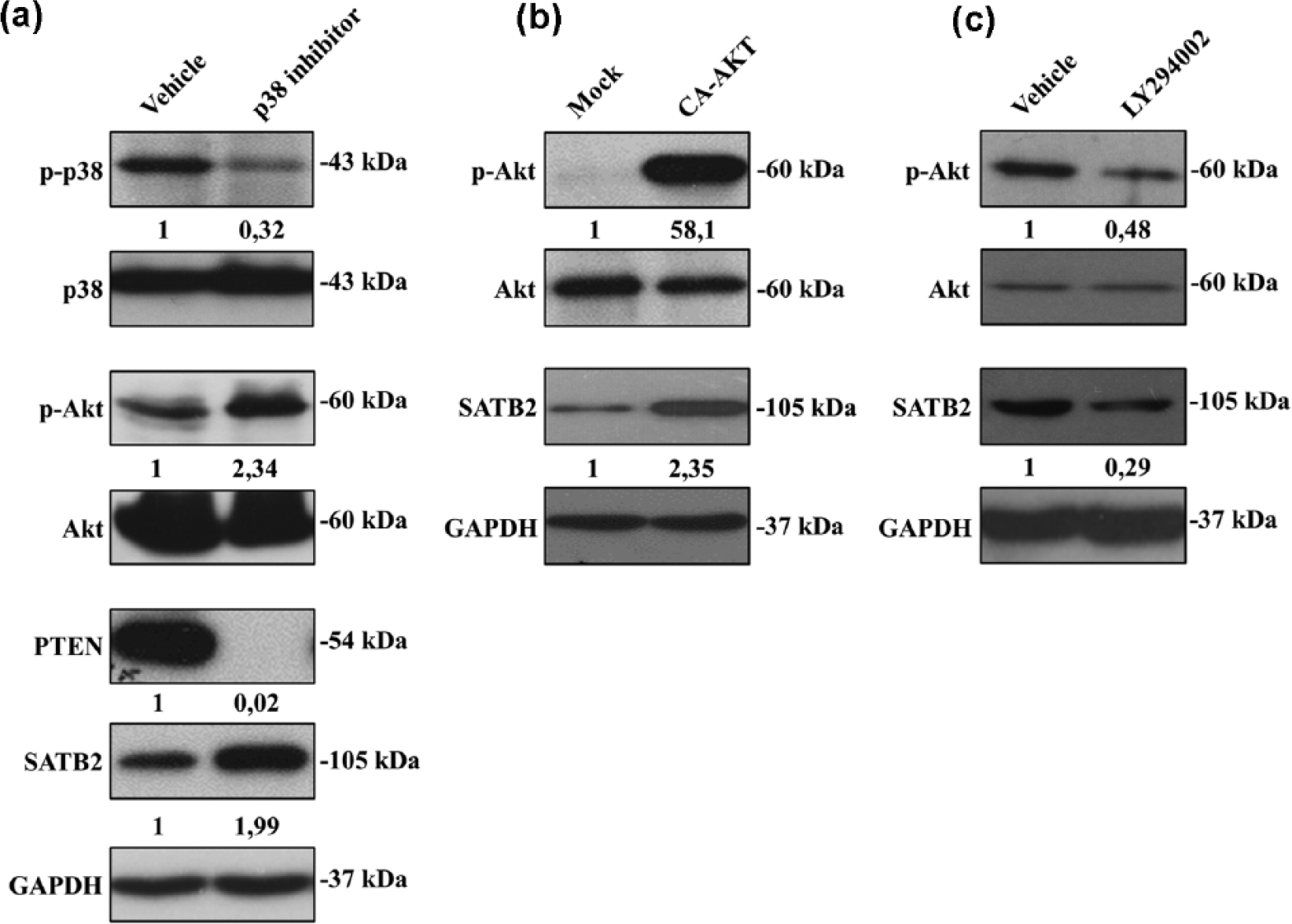
Inhibition of p38 leads to increased Akt activation and upregulation of SATB2 expression. (a) A549 cells were treated with SB203580 (500 nM) or DMSO in serum-free medium for 24 h. Cellular lysates were prepared in RIPA buffer and equal amount of proteins was fractionated using 10% polyacrylamide gel, and blots were labeled with p-p38-, p38-, p-Akt-, Akt-, PTEN-, SATB2-, and GAPDH-specific antibodies. (b) A549 cells were transfected with continuous active Akt (CA-Akt) vector or mock vector for 24 h. Cellular lysates were prepared in RIPA buffer and equal amount of proteins were fractionated using 10% polyacrylamide gel; blots were labeled with p-Akt-, Akt-, SATB2-, and GAPDH-specific antibodies. (c) A549 cells treated with LY294002 (20 nM) or DMSO in serum-free medium for 24 h. Cellular lysates were prepared in RIPA buffer and equal amount of proteins was fractionated using 10% polyacrylamide gel, and blots were labeled with p-Akt-, Akt-, SATB2-, and GAPDH-specific antibodies. The band intensities on films were quantified by densitometric analysis using Image Studio Lite Ver 5.2, and values of bands were normalized against p38 or Akt or GAPDH. To calculate fold inductions, the expression values of bands were divided by that of controls.
p38 activation is required for maintenance of mesenchymal phenotype of NSCLC cells
In our recently published study, it has been showed that low expression of SATB2 is critical for acquisition of mesenchymal phenotype in NSCLC cells. 10 Furthermore, our results clearly indicated that there is a potent association between the expression of SATB2 and E-cadherin, and p38 inhibition strongly induces SATB2 expression of NSCLC cells in this study (Figure 1(b)). Therefore, we wanted to examine the effects of p38 inhibition on expression of epithelial and mesenchymal markers in A549 cells. As shown in Figure 3(a), suppression of p38 activation by SB203580 resulted in dramatic reduction and induction of E-cadherin, N-cadherin, and vimentin in A549 cells. After showing the effect of p38 inhibition on EMT markers, we determined the protein levels of EMT-associated transcription factors Snail, Slug, Zeb1, and Twist1 in the same lysate, and we found that p38 inhibition causes significant induction of the protein levels of Snail and Twist1 and dramatic downregulation in that of Slug and Zeb1. These results clearly demonstrate that p38 activation has an important role in the maintenance of mesenchymal phenotype by regulating the expressions of EMT-associated transcription factors in NSCLC cells. Next, we wondered whether p38 inhibition would interfere with capacities of migration and invasion of A549 cells. Therefore, wound-healing and invasion assays were performed to measure the effects of p38 inhibition on migration and invasion of A549 cells. As shown in Figure 3(c), wound size was photographed at 0, 16, and 24 h. After 16 and 24 h of SB203580 treatment, p38 inhibition decreased migration capacity of A549 cells compared to untreated control group. p38 inhibition also caused to decrease invasion capacity of A549 cells at the rate of 40% (Figure 3(d)). These results clearly indicate that p38 activation plays a critical role in migration and invasion of NSCLC cells. Furthermore, to confirm the possibility of cell death induced by SB203580 treatment affects on cellular migration and invasion, we performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Interestingly, we observed that p38 inhibition by SB203580 treatment in A549 cells leads to the increase in cell proliferation at the rate of 19% (Figure 3(e)).
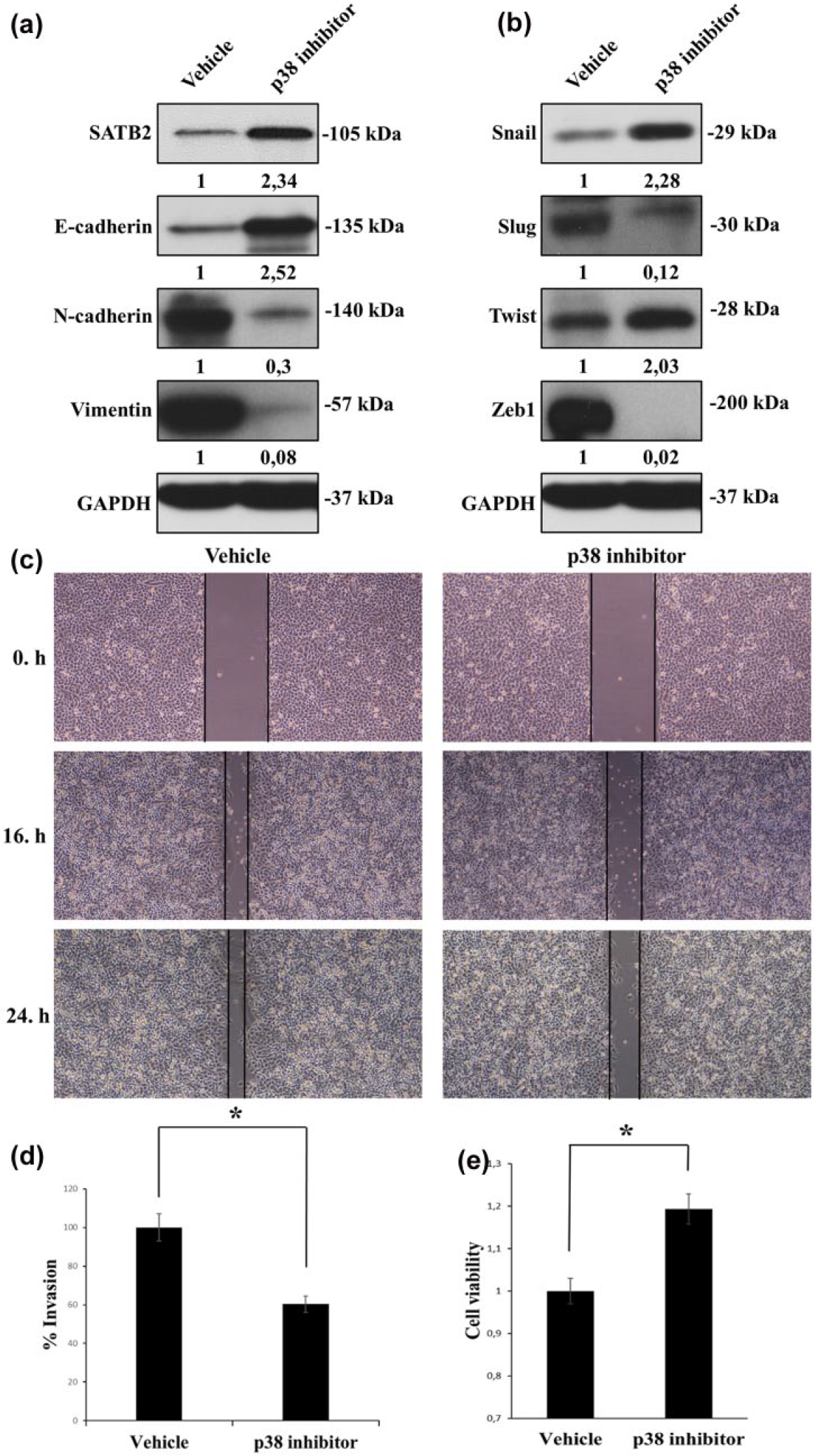
p38 activation is required for maintenance of mesenchymal phenotype of NSCLC cells. (a) A549 cells were treated with SB203580 (500 nM) or DMSO in serum-free medium for 24 h. Cellular lysates were prepared in RIPA buffer and equal amount of proteins were fractionated using 10% polyacrylamide gel, and blots were labeled with SATB2-, E-cadherin-, N-cadherin-, vimentin-, and GAPDH-specific antibodies. (b) The same cellular lysates were fractionated using 10% polyacrylamide gel, and blots were labeled with Snail-, Twist-, Slug-, Zeb1-, and GAPDH-specific antibodies. The band intensities on films were quantified by densitometric analysis using Image Studio Lite Ver 5.2, and values of bands were normalized against p38 or Akt or GAPDH. To calculate fold inductions, the expression values of bands were divided by those of controls. (c) A549 cells seeded into six-well plates at 80%–90% confluency. The wound was scratched using 200 μL pipette tip. To remove cell debris, the cells were washed with serum-free medium for three times, and the cells were treated with SB203580 (500 nM) or DMSO in serum-free medium. Wound size at 0, 16, and 24 h was photographed under the phase-contrast microscope with 20× magnification. (d) A549 cells were trypsinized and seeded into Matrigel-coated Transwell chambers and treated with SB203580 (500 nM) or left DMSO for 24 h in serum medium. At the end of incubation time, invading cells were fixed, stained, and counted from the independent field per chamber (*p < 0.05). (e) A549 cells were seeded into 96-well tissue culture plates, and the cells were treated with p38 inhibitor SB203580 (500 nM) or DMSO for 24 h and cell viability was measured using Vybrant® MTT Cell Proliferation Assay Kit according to the manufacturer’s instructions. Formazan formation was quantified spectrophotometrically at 560 nM wavelength using a microplate reader (*p < 0.05).
SATB2 knockdown prevented the effect of p38 inhibition on EMT in A549 cells
Since p38 inhibition reversed EMT in A549 cells via upregulation of SATB2 expression, we wondered whether SATB2 downregulation can prevent the effect of p38 inhibition on EMT in A549 cells. To answer this question, we first transfected A549 cells with SATB2 siRNA or non-targeting control siRNA and observed the expression of SATB2 and E-cadherin. SATB2 knockdown alone resulted in downregulation of E-cadherin in A549 cells (Figure 4(a)). Next, we treated SATB2-knockdowned A549 cells with SB203580 for 24 h. As shown in Figure 4(b), siRNA-mediated knockdown of SATB2 abolished increased E-cadherin expression induced by p38 inhibition and prevented downregulations of N-cadherin and Vimentin via p38 inhibition as well. These results first showed that p38 controls EMT process by regulating SATB2 expression in NSCLC cells. After showing the role of SATB2 expression in effects of p38 inhibition on EMT, we determined the protein levels of EMT-associated transcription factors Slug and Zeb1 in the same lysate and found that SATB2 knockdown abrogated downregulations of Slug and Zeb1 induced by p38 inhibition (Figure 4(c)).

SATB2 knockdown prevented the effect of p38 inhibition on EMT in A549 cells. (a) A549 cells were transfected with SATB2 siRNA or non-targeting control siRNA (15 nM) for 24 h, cellular lysates were prepared in RIPA buffer and equal amount of proteins were fractionated using 10% polyacrylamide gel, and blots were labeled with SATB2-, E-cadherin-, and GAPDH-specific antibodies. (b) A549 cells were transfected with SATB2 siRNA or non-targeting control siRNA (15 nM) for 24 h. Then, cells were treated with SB203580 (500 nM) or DMSO in serum-free medium and cellular lysates were prepared in RIPA buffer and equal amount of protein was fractionated using 10% polyacrylamide gel; blots were labeled with E-cadherin-, N-cadherin-, vimentin-, and GAPDH-specific antibodies. (c) The same cellular lysates were fractionated using 10% polyacrylamide gel, and blots were labeled with Slug-, Zeb1-, and GAPDH-specific antibodies. The band intensities on films were quantified by densitometric analysis using Image Studio Lite Ver 5.2, and values of bands were normalized against GAPDH. To calculate fold inductions, the expression values of bands were divided by that of controls.
Discussion
EMT is a both physiological and pathological process and plays a crucial role in biological events such as embryonic development, tissue repair, fibrosis, and carcinogenesis. Normally, epithelial cells have a tight cell–cell junctions, apico-basal polarity, and weak migratory ability. During EMT process, it enables epithelial cells to acquire mesenchymal cell traits, including weakened cell–cell junctions, loss of apico-basal polarity, and increased migratory ability.1,11,12 Since EMT is a complex event, and controlled by a wide variety of transcription factors and signaling pathways, it is not surprising that the crosstalks between this signaling pathways could implicate in regulation of EMT process. Emerging evidences have recently demonstrated that crosstalks between several pathways play a critical role in the regulation of EMT.11,13–15 However, underlying mechanisms of the crosstalks between several signaling pathways involved in EMT are still unknown. It is well known that there is a crosstalk between p38 and Akt signaling pathways, and this crosstalk is involved in physiological processes such as myoblast differentiation.8,9,16,17 Since it has not been reported that whether the crosstalk between p38 and Akt signaling pathways is involved in the regulation of EMT, we aimed to illuminate the effects of this crosstalk on the regulation of EMT process in NSCLC cells in this study.
In our recently published study, we showed that downregulation of SATB2 is critical for induction of EMT and invasion of NSCLC cells. 10 Therefore, we first examined SATB2 expression as well as E-cadherin expression to evaluate epithelial-mesenchymal character of A549 and H1650 NSCLC cells, and these cells were used as a model in our study. As shown in Figure 1(b), E-cadherin and SATB2 are upregulated in H1650 cells compared to A549 cells. To determine whether there is a crosstalk between p38 and Akt pathways in these cells, we analyzed the expression levels of p-p38, p38, p-Akt, and Akt in normal living conditions of these cells. Consequently, we first showed that there is the crosstalk between p38 and Akt signaling pathway in NSCLC cells. These results are in good agreement with the publications which showed that there is this crosstalk in several cell lines.8,9 Then, we hypothesized that this crosstalk might determine epithelial-mesenchymal character of NSCLC cells such as A549 and H1650 cell lines that have a distinct epithelial character.
Previous studies showed that p38 has a regulatory effect on Akt signaling pathway by modulating PTEN expression.9,16 Thus, we thought to interfere the crosstalk between p38 and Akt pathways with inactivation of p38 pathway via SB203580 to determine the effects of this crosstalk on NSCLC cells. As expected, p38 inhibition resulted in decreased PTEN expression and subsequently increased Akt activation in NSCLC cells (Figure 2(a)). These results are in good agreement with the publications which showed that p38 has a regulatory role on Akt pathways by modulating PTEN expression.9,16–18 Previous study showed that inhibition of p38 blocks the upregulation of Satb2 in sympathetic neuron cells during cholinergic transdifferentiation. 19 However, we found that p38 inhibition strongly induced SATB2 expression in NSCLC cells. It has not been reported that there is the crosstalk between p38 and Akt pathways in sympathetic neuron cells. Therefore, lack of this crosstalk in sympathetic neuron cells may be a reason of the diversity in the association between p38 activation and SATB2 expression. Furthermore, we wondered whether activation or inactivation of Akt could regulate SATB2 expression in NSCLC cells. It was observed that enhanced activation of Akt induced SATB2 expression while inactivation of Akt pathway induced by LY294002 leads to downregulation of SATB2 expression, indicating that SATB2 expression is tightly regulated by Akt in NSCLC cells.
It has been described that p38 activation is required for the induction of EMT in epithelial cells.1,3,20–22 Under the light of these, p38 inhibition in A549 cells is logical to evaluate whether the crosstalk between p38 and Akt pathways has a regulatory role in EMT of NSCLC. As shown in Figure 3(a), just like we anticipated, we observed that p38 inhibition dramatically reversed EMT in NSCLC cells by upregulating SATB2 expression. Previously, we reported that SATB2 seems to be responsible for suppression of expression of these EMT-related transcription factors. 10 We found that p38 inhibition resulted in the obvious downregulations of Slug and Zeb1, related to especially EMT in NSCLC. However, p38 inhibition upregulated expressions of Snail and Twist. It is known that Snail and Twist are positively regulated by Akt signaling pathway.23–26 Therefore, increased activation of Akt via p38 inhibition might induce expression of Snail and Twist in NSCLC cells. Moreover, p38 inhibition decreased migration and invasion of NSCLC while increased NSCLC proliferation (Figure 3(c)–(e)). It is well known that cancer cells display a phenotype with decreased proliferation and increased capacities of migration and invasion during EMT.27–30 Thus, the reversion of EMT induced by inhibition of p38 may be a reason to explain why p38 inhibition enhances cell proliferation in NSCLC cells. Finally, we wondered whether SATB2 downregulation could prevent the effect of p38 inhibition on EMT in NSCLC cells. Here, we first report that SATB2 knockdown abrogated the effect of p38 inhibition on EMT in NSCLC cells via especially Slug expression in this study (Figure 4(a) and (b)).
In conclusion, these results strongly indicate that SATB2 has a key importance in EMT regulation by the crosstalk between p38 and Akt pathways in NSCLC cells. Our findings will contribute to illuminate the molecular mechanisms of the EMT process that has a critical significance for lung cancer metastasis. Consequently, these results should be considered when using p38 and Akt inhibitors as anticancer drugs in lung cancer therapy.
Footnotes
Acknowledgements
The authors would like to thank Dr Ozes for CA-Akt overexpression vector.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants (114S007 and 215Z283) from TUBITAK.