
Abstract
As a transcription factor, p53 must accumulate in the nucleus to be effective. Signals related to nuclear localization are distributed mainly in the C-terminal of p53. So these nuclear location domains were reserved and the other part of the C-terminal was removed in this study. We investigated whether the truncated p53 (p53(DEL)) may affect proliferation and invasive potential of human lung cancer cells. H1299 and 801D cells expressing full-length p53 and the p53(DEL) were obtained by screening. Cell proliferation assay, cell apoptotic analysis, cell migration assay, and invasion assay were performed. Protein expression was examined by Western blotting. The data showed H1299-p53(DEL) and 801D-p53(DEL) cells grew more slowly than H1299-p53 and 801D-p53 cells, respectively. The colony formation of H1299-p53(DEL) and 801D-p53(DEL) cells reduced. The truncated p53 induced cell apoptosis. The expression levels of Bax and p53 upregulated modulator of apoptosis were increased in H1299-p53(DEL) and 801D-p53(DEL) cells. H1299-p53(DEL) and 801D-p53(DEL) cells were also characterized by decreased migration and invasion. The expression of the truncated p53 resulted in upregulation of E-cadherin and downregulation of Vimentin, Slug, Twist1, and zinc finger E-box-binding homeobox 1, which suggested the truncated p53 inhibited epithelial–mesenchymal transition occurrence. The above-mentioned characteristics were reverted by treatment of with pifithrin-a, a p53 inhibitor. These findings support the existence of a direct link between the p53(DEL), proliferation, epithelial–mesenchymal transition, and invasiveness in human lung cancer cells. So the p53(DEL) is a promising target for prevention and treatment of lung cancer.
Introduction
Lung cancer is a genetic disease that can take decades to develop through the accumulation of genetic mutations that activate oncogenes and inactivate tumor suppressor genes. Tumor suppressors act as “cellular guardians,” by inhibiting cell growth pathways and/or by inducing cellular apoptosis, consequently preventing cancer formation. 1 p53 is a key tumor suppressor, also known as the guardian of the genome.2,3 p53 was shown to exert its tumor suppressive functions by inducing cell cycle arrest, apoptosis, DNA repair, and more.4,5 If p53 is dysfunctional or deficient, it can result in genomic instability or escape apoptotic pathways and proliferate indefinitely under the selection of cellular stresses. Thus, p53 plays a key role in the prevention of carcinogenesis.3,5,6
p53 is mainly composed of N-terminal transactivation domain, central DNA-binding domain, and C-terminal functional domain. The C-terminal of p53 includes nuclear localization signal (NLS), nuclear export signal (NES), tetramerization domain, and C-terminal regulatory domain. As a transcription factor, p53 must accumulate in the nucleus to be effective.7–9 Therefore, NLS and NES are essential for the function of p53. The tetramerization domain is essential for DNA binding, protein–protein interactions, post-translational modifications, and p53 degradation.10,11 So we speculate that NLS, NES, and the tetramerization domain of the C-terminal are essential for its function and the other part of the C-terminal may not play an important role. Thus, these nuclear location domains were reserved and the other part of the C-terminal was removed in this study. To confirm the assumption, we carried out the related study about the function of the truncated p53 in lung cancer cells. The results showed that the truncated p53 could better inhibit cell proliferation and weaken cell invasion ability in comparison with the full-length p53 in lung cancer cells. In addition, the truncated p53 induced cell apoptosis and inhibited epithelial–mesenchymal transition (EMT) occurrence in lung cancer cells. The above roles were reversed by treating with pifithrin-a (PFT-a), a p53 inhibitor.
Materials and methods
Cell lines and culture conditions
H1299 (human lung adenocarcinoma cell line) and 801D (human large cell lung carcinoma cell line) were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Los Angeles, CA, USA). The cells were maintained at 37°C in a humidified chamber containing 5% CO2 and 95% air.
p53 gene status in human lung cell lines and p53 plasmid transfection
801D and H1299 cells were selected. The p53 gene exhibits loss of heterozygosity in the 248th codon and a CGG → CTT transversion in 801D cells and deletion in H1299 cells. The two cell lines were used as target cells for p53 transfection. For stable transfection, the cells were seeded at 106 cells per 6-cm Petri dish 24 h before transfection. Three plasmids used for the study were pEGFP, pEGFP-containing p53 complementary DNA (cDNA), and pEGFP-containing p53 cDNA with partial C-terminal deletion (amino acids 357–393; Figure 1). The plasmid of pEGFP-containing p53 cDNA with partial C-terminal deletion (amino acids 357–393) was named as p53(DEL). A volume of 5.0 × 105 801D and 3.0 × 105 H1299 cells were seeded on 35 mm plates. The two plasmids were transfected into cells using lipofectamine (Lipofectamine 2000; Promega, Madison, WI, USA). The final concentration of the plasmid was 1 µg/220 µL. G418 (Gibco, Gaithersburg, MD, USA) was added to a final concentration of 800 µg/mL at 72 h after transfection. After 2–3 weeks of G418 selection, resistant cells were then cloned by limiting dilution. The cell lines transfected with p53 cDNA and p53 cDNA with partial C-terminal deletion were named 801D-p53 and 801D-p53(DEL) in 801D cells and H1299-p53 and H1299-p53(DEL) in H1299 cells, respectively. Control-transfected strains were named 801D-p in 801D cells and H1299-p in H1299 cells, respectively.

Structure diagrams of the truncated p53 and wild-type p53.
Cell proliferation assay
Tumor cells (3000/well) were seeded in flat-bottom 96-well plates. Cell proliferation was evaluated by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Promega) assay, which was performed at a fixed time every day for the next 5 days. A total of 20 µL of MTS was added to each well, followed by incubation for 3 h. The absorbance was recorded at 490 nm with an EL-800 Universal Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA). This assay was repeated three times.
Colony formation assay
A total of 300 cells were suspended in 2 mL of culture medium and seeded on six-well plates. The cells were maintained for 10 days with a change of media every 3–4 days. The number of colonies with >50 cells in each well was counted on the 10th day. The colonies were visualized and counted by the trypan blue exclusion method. The assay was repeated three times.
Cell apoptotic analysis
Cells were seeded on 96-well plates and grown to 75% confluence. Cells in logarithmic growth phase were collected. After washing with RPMI-1640 medium without serum, cells were incubated in 100 µL RPMI-1640 medium without serum (50 mg/mL propidium iodide (P4170; Sigma-Aldrich, St. Louis, USA) and 5 µg/mL Hoechst 33342 (R31165; Thermo Fisher Scientific, Waltham, USA). Samples were incubated for 10 min at 37°C, followed by determination of the percentage of apoptotic cells by Thermo Scientific ArrayScan VTI HCS (Thermo Fisher Scientific). CellQuest software (Becton Dickinson, NJ, USA) was used for analysis.
Cell migration assay
Cell motility was assessed by two assays. For the wound-healing assay, confluent monolayer cells were wounded with a sterile pipette tip and cultured in serum-free medium on six-well plates. The wounds were observed at 0, 12, and 24 h along the scratch, and representative images of fixed positions were acquired with a phase-contrast microscope. The wound areas were measured using Alpha View Analysis Tools, and the percentage wound closure was determined.
A migration assay was performed in a 24-well Transwell unit containing an 8 µm pore size polycarbonate membrane (Costar, Cambridge, NY, USA) as reported previously. After starvation for 12 h, the cells were suspended and plated on the upper compartment with serum-free medium. The lower compartment was filled with medium containing 10% FBS that was used as a chemoattractant. After 24 h, the cells in the upper compartment were removed completely by gentle swabbing. Cells migrating to the lower surface of the membrane were determined using crystal violet staining. The number of cells on the lower surface of the membrane was counted in five microscopic fields at 200× magnification. Triplicate samples were tested. The data are presented as mean ± standard error of the mean (SEM).
Invasion assay
Cell invasion was analyzed by employing Boyden chambers equipped with 8 µm porosity polyvinylpyrrolidone-free polycarbonate filters that were coated with 5 µg/mL of matrigel solution (BD Pharmingen, San Diego, CA, USA). The filters were stained with crystal violet solution. Cell invasion was quantified by counting, with a Zeiss microscope (Oberkochen, Germany) equipped with bright-field optics (final magnification: 400×), crystal violet-stained cells that invaded matrigel. For each filter, cells in 10 randomly chosen fields were counted and expressed as the number of invading cells per high-power field. The data are presented as mean ± SEM.
Study on malignant phenotype of cells after using p53 inhibitor, PFT-a
Cell proliferation assay, migration assay, and invasion assay were also assessed in the presence of p53 inhibitor PFT-a (s2929; Selleck Chemicals, Houston, TX, USA). In brief, H1299 and 801D cells were seeded onto 96-well plates or 6-well plates. PFT-a (0, 20, 50, and 100 µM) was added 24 h later. The vehicle (dimethyl sulfoxide (DMSO); 0.3 µL/mL) was used as a control.
Western blotting
Protein samples of 50–100 mg were subjected to 12% polyacrylamide gel electrophoresis. The separated proteins were transferred to a polyvinylidene difluoride membrane by electrotransfer. The blots were blocked with 5% milk (Protifar; Nutricia, Zoetermeer, The Netherlands) in Tris-buffered saline with Tween 20 (TBST: 10 mM Tris, pH 8.0; 150 mM NaCl; and 0.001% Tween 20) at room temperature for 1 h and incubated overnight at 4°C with specific antibodies against p53 (sc-47698; 1:1000; Santa Cruz Biotechnology, CA, USA), Lamin B1 (sc-56145; 1:1000; Santa Cruz, CA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; kc-5G5; 1:3000; Kandcheng, Shanghai, China) diluted in 5% milk/TBST.
For the detection of zinc finger E-box-binding homeobox 1 (ZEB1), Vimentin, Slug, E-cadherin, and Twist1, mouse anti-ZEB1 antibody (Abcam, Cambridge, UK), mouse anti-Vimentin antibody (Abcam), mouse anti-Slug antibody (Abcam), rabbit anti-E-cadherin antibody (Abcam), mouse anti-Twist1 antibody (Abcam), rabbit anti-Bax antibody (Abcam), and rabbit anti-PUMA antibody (Abcam) were used.
Cytosol and nuclear fractions
801D and H1299 cell lines were collected and washed twice with ice-cold phosphate-buffered saline (PBS) and pelleted by centrifugation at 500g for 5 min. A volume for 1–10 × 106 cells were transferred to a 1.5 mL microcentrifuge tube and pelleted by centrifugation at 500g for 2–3 min. We used “NE-PER Nuclear and Cytoplasmic Extraction Reagents” Kit (78840; Pierce, Waltham, USA) for step-by-step extraction of the cytoplasmic and nuclear proteins. Protein concentration in both the cytoplasmic and nuclear fractions was estimated by the Bradford method (5000001; Bio-Rad Protein Assay, Hercules, USA).
Statistical analysis
The data are presented as the mean ± SEM of at least three independent experiments. Statistical analysis was performed using one-way analysis of variance (ANOVA) or Student’s t test, using SPSS software (SPSS Inc., Chicago, IL, USA). Unless otherwise indicated, p < 0.05 was deemed significant.
Results
p53 expression in the transfected cell lines
For detection of p53, 50 mg of protein of human lung cancer cell lines was subjected to Western blotting (Figure 2(a) and (b)). Compared with 801D and 801D-p cells, p53 protein levels of 801D-p53 and 801D-p53(DEL) cells increased. Compared with H1299 and H1299-p cells, p53 protein levels of H1299-p53 and H1299-p53(DEL) cells increased under standard culture conditions. p53 expression was obviously reduced by 89.5% and 87.5% in 801D and H1299-p53 cells respectively when 100 µM PFT-a was used (Figure 2(c) and (d)).

Expression of p53 in lung cancer cell lines. (a) Expression of p53 in 801D cells and their transfected cell strains. (b) Expression of p53 in H1299 cells and their transfected cell strains. (c) Expression of p53 in 801D cells using different concentrations of PFT-a. (d) Expression of p53 in H1299 cells using different concentrations PFT-a.
The truncated p53 better suppressed cell proliferation than the full-length p53
To determine the function of the truncated p53, we performed cell proliferation assay and colony formation assay. As indicated in Figure 3(a), 801D-p53 and 801D-p53(DEL) cells decreased growth on the second day. Up to day 3, proliferation of cells with full-length p53 and with p53(DEL) was dramatically slower than that of the parent cells. Notably, proliferation of cells with p53(DEL) was slower than that of the cells with full-length p53 from the third day. To determine whether p53(DEL) plays a role in cell proliferation, 801D and H1299 cells were incubated with p53 inhibitor PFT-a at a concentration of 100 µM. MTS assay was performed. As shown in Figure 3(b), the proliferation ability of 801D-p53(DEL) and H1299-p53(DEL) was almost the same as that of 801D-p53, H1299-p53, and the parallel cells after the treatment with PFT-a.

Effect of p53(DEL) on tumorigenesis. (a) p53(DEL) inhibited the proliferation of 801D and H1299 cells. OD490 values were determined daily at predetermined time points. The data are expressed as mean of triplicate values from three separate experiments (*p53(DEL) compares with the controls, **p53(DEL) compares with the control and full-length p53). (b) The effect of PFT-a on cell proliferation was detected by MTS assay. The data are expressed as mean. (c) p53(DEL) obtained density-dependent growth inhibition. Colony formation after 10 days was quantified by counting the number of colonies per well. The values represent the mean number of colonies ± SD from three separate wells (*p53(DEL) compares with the controls, **p53(DEL) compares with the control and full-length p53). (d) The effect of PFT-a on cell density–dependent growth inhibition. The data are expressed as mean.
The apoptosis ratio increased in cells with the truncated p53
Induction of apoptosis was analyzed by calculating the apoptosis ratios by HCS after the cells were treated with or without PFT-a. The cells with p53(DEL) and with full-length p53 appeared to undergo more apoptosis than the controls (801D and H1299; Table 1). The cells with p53(DEL) appeared to undergo more apoptosis than the cells with full-length p53. In 801D-p53 and H1299-p53 cells, the apoptosis ratios versus the controls increased 1.99- and 2.64-fold, respectively (p < 0.05). In 801D-p53(DEL) and H1299-p53(DEL) cells, the apoptosis ratios versus the controls increased 2.65- and 4.05-fold, respectively (p < 0.05). In cells with p53(DEL) and cells with full-length p53, the expression levels of Bax and PUMA (p53 upregulated modulator of apoptosis) are significantly increased with respect to control cells (Figure 6(a) and (b)). These protein levels in cells with p53(DEL) were not different with cells with full-length p53. The apoptosis rates of cells with p53(DEL) and with full-length p53 were obviously reduced by the treatment of PFT-a.
Apoptosis ratio in human lung cancer cell lines.

PFT-a: pifithrin-a.
compared with the control; *Δcompared with cells with the control and full-length p53.
The truncated p53 better inhibits lung cancer cell tumorigenesis than the full-length p53
To gain additional insight into the effect of p53(DEL) on lung cancer cells growth, cell tumorigenesis was assessed by colony formation assays. The result indicated that the colony-forming efficiency of cells with p53(DEL) and cells with full-length p53 was less than half that of the parallel cells (Figure 3(c)). Moreover, the colony-forming efficiency of cells with p53(DEL) was lower than that of cells with full-length p53. Thus, p53(DEL) may obtain more density-dependent growth inhibition than full-length p53 in lung cancer cells. The growth of 801D-p53(DEL) and H1299-p53(DEL) cells was not different with that of 801D-p53, H1299-p53 and the controls by the treatment of PFT-a (Figure 3(d)).
The truncated p53 better decreases the migration and invasion ability than the full-length p53 in non–small cell lung cancer cells
Migration and invasion are important characteristics of malignant cancer cells. So we investigated the effect of p53(DEL) on cell migration and invasion. In the invasion assay, none of the cells were observed when cells were incubated for 24 h, and we counted the number of cells on the lower surface of the membrane when cells were incubated for 48 h. Both cell migration, as determined using wound-healing and Transwell assays (Figure 4(a) and (b)), and cell invasion (Figure 4(c)) were inhibited in cells with p53(DEL) and cells with full-length p53 compared to the corresponding controls. To be sure, cell migration and invasion were better or similarly inhibited in cells with p53(DEL) than cells with full-length p53. These findings indicate that p53(DEL) could better inhibit the migration and invasion ability of lung cancer cell than full-length p53. The ability of p53(DEL) on invasion ability had been restored by the treatment of PFT-a (Figure 5(a)–(c)).

The rates of migration and invasion in cells with p53(DEL). (a) Migration was investigated using a wound-healing assay. The percent areas of wound closure at 24 h following wound generation were calculated from three separate areas and are expressed as the mean. Photomicrographs were taken after 24 h (original magnification: 100×). (b) Other migration assays were performed using a Transwell unit. Cells on the underside of the insert filters were fixed, stained, and counted under a microscope. The data are expressed as mean of triplicate values from three separate experiments. Photomicrographs were taken after 24 h (original magnification: 200×). (c) The in vitro invasive properties of cells were tested using a matrigel-coated Transwell unit. Cells on the underside of the insert filters were fixed, stained, and counted under a microscope. The data are expressed as mean ± SEM of triplicate values from three separate experiments. Photomicrographs were taken after 48 h (original magnification: 400×).

The rates of migration and invasion in cells with PFT-a. (a) The effect of PFT-a on cell migration was investigated using a wound-healing assay. (b) The effect of PFT-a on cell migration was performed using a Transwell unit. (c) The effect of PFT-a on cell invasiveness was performed using a matrigel-coated Transwell unit.
The truncated p53 correlates with the downregulation of EMT biomarkers
The morphological changes characteristic of cells undergoing EMT are accompanied by a shift in gene expression from an epithelial to a mesenchymal repertoire. To determine whether p53(DEL) inhibits migration and invasion of lung cancer cells, both the expression and cellular distribution of selected EMT markers have been investigated by Western blotting. In cells with p53(DEL) and cells with full-length p53, Slug, Twist1, ZEB1, and Vimentin protein levels are significantly diminished and E-cadherin protein levels are upregulated with respect to control cells (Figure 6(a) and (b)). These protein levels in cells with p53(DEL) were not different with cells with full-length p53. These protein levels in cells with p53(DEL) and cells with full-length p53 increased but not return to the levels of the parallel cells by the treatment of PFT-a (Figure 6(c) and (d)).
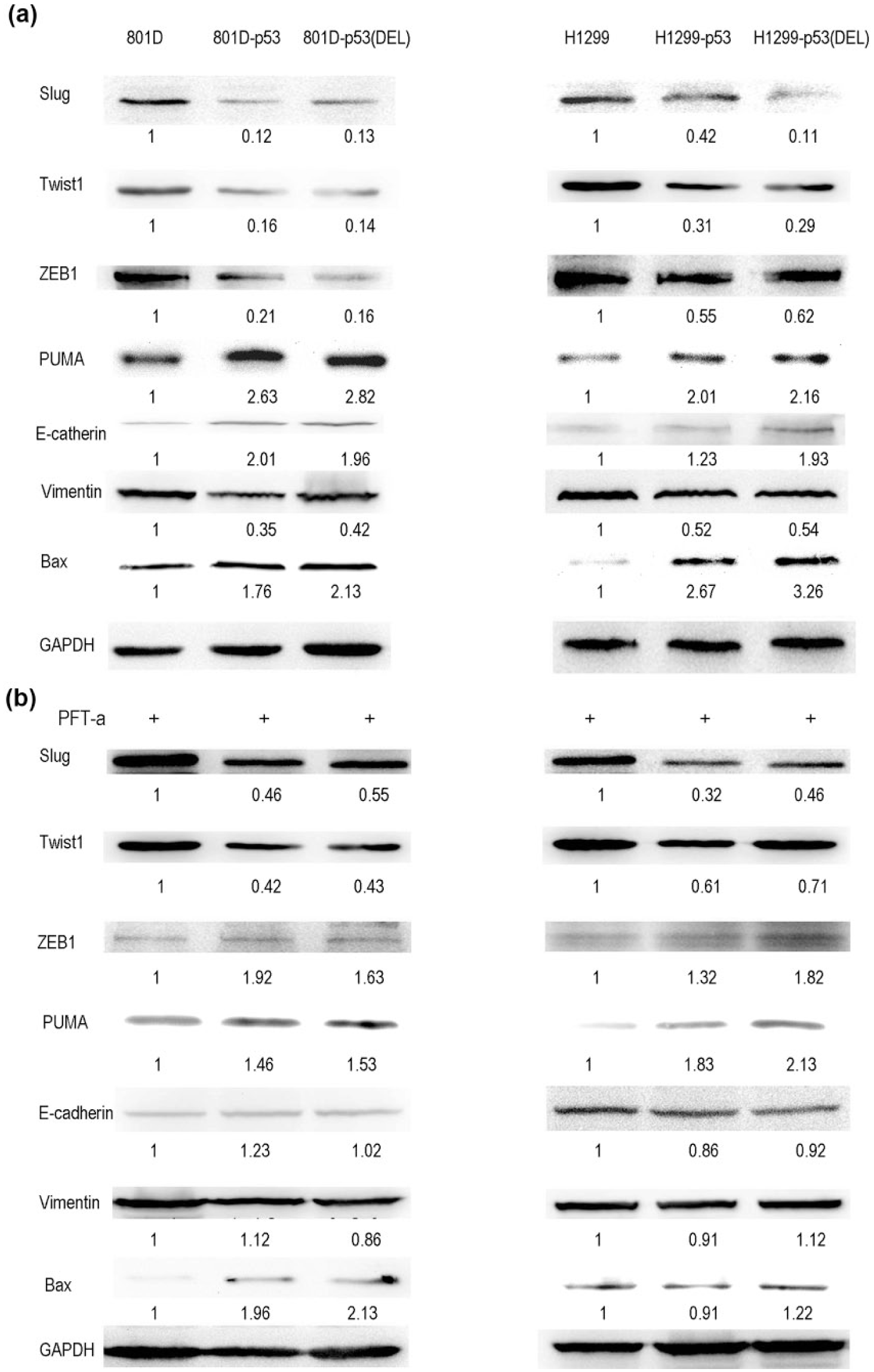
Expression of apoptosis and EMT markers in cells with p53(DEL). Total cell lysates were analyzed by Western blotting to determine the expression levels of Bax, PUMA, E-cadherin, ZEB1, Vimentin, Twist1, and Slug. The optical density of the bands was determined by densitometry, normalized with respect to that of the corresponding GAPDH band and expressed as arbitrary units relative to the control (SD < 10%) in (a) 801D cells, (b) H1299 cells, (c) 801D cells + PFT-a, and (d) H1299 cells + PFT-a.
Active p53 expression increased in the truncated p53 cells
As a transcription factor, nuclear localization of p53 is essential for its role in tumor suppression. So we studied subcellular localization of p53. As well as in 801D-p53 and H1299-p53 cells, nuclear levels of p53 were increased in 801D-p53(DEL) (Figure 7(a)) and H1299-p53(DEL) (Figure 7(b)). By contrast, nuclear levels of p53 in 801D-p53(DEL) were lower than that of p53 in 801D-p53 and nuclear levels of p53 in H1299-p53(DEL) were lower than that of p53 in H1299-p53.

The nuclear/cytosol ratios of p53 in two lung cancer cell lines and their transfected cell strains (a) 801D cells and (b) H1299 cells.
Discussion
In p53(DEL), we reserved NLS, NES, and tetramerization domains and removed the other part of the C-terminal. Is the function of p53(DEL) is similar to wild-type p53 in lung cancer cells? So we studied the effect of p53(DEL) on proliferation ability of lung cancer cells. Data showed that p53(DEL) better suppressed cell proliferation than the full-length p53 in 801D and H1299 cells. But the growth difference was displayed in the fourth day in 801D-p53(DEL) cells and in the third day in H1299-p53(DEL) cells. Studies show that mutant p53 can acquire new functions to drive cell migration, invasion, and metastasis.12–17 So mutant p53 can partly offset the function of wide-type p53, and the function of wide-type p53 is delayed as shown in 801D cells with mutated p53. In H1299 cells with deleted p53, the exogenously introduced p53 exerts the function quickly. In addition, p53(DEL) induces more cell apoptosis than wild-type p53, but obvious cell cycle arrest does not appear in these cells (the data are not shown). In addition, the expression levels of Bax and PUMA in 801D-p53(DEL) and H1299-p53(DEL) cells are significantly increased with respect to control cells. This suggests that p53(DEL) can induce apoptosis in these cells.
p53 not only prevents tumorigenesis but also tumor progression, that is, local invasion and distant metastasis.18–23 So we hypothesize that p53(DEL) can affect migration and invasion ability of lung cancer cells. Our data show that 801D-p53 and H1299-p53 cells could better pass through the matrigel matrix than 801D-p53(DEL) and H1299-p53(DEL) cells. This effect on invasion ability can be inhibited by PFT-a in lung cancer cells. Previous studies showed that p53 could reduce Slug,24–26 Twist1,23,27–29 ZEB1,23,30,31 and Vimentin23,32 expression levels and enhance E-cadherin33–35 expression. Then, whether p53(DEL) may affect EMT? In this study, Slug, Twist1, ZEB1, and Vimentin levels of acquired EMT markers are reduced and E-cadherin levels of attenuated EMT markers increased in 801D-p53(DEL) and H1299-p53(DEL) cells the same as 801D-p53 and H1299-p53 cells. These marker levels can wholly or partially be recovered by PFT-a. The data showed that p53(DEL) decreases cell migration and invasion and inhibits EMT occurrence in non–small cell lung cancer (NSCLC) cells.
p53 needs to translocate to the nucleus to activate endogenous target genes. Thus, nuclear localization of p53 is essential for its function as a transcription factor. 36 In this study, nuclear levels of p53 increase in 801D-p53(DEL) and H1299-p53(DEL) cells the same as 801D-p53 and H1299-p53 cells, compared with the parallel cells. But nuclear levels of p53 are lower in 801D-p53(DEL) and H1299-p53(DEL) cells than 801D-p53 and H1299-p53 cells. The reason is that p53(DEL) removes two minor NLS (370–376 and 380–386). Studies have reported that amino acids 305–322 (NLS) and the C-terminal basic domain (amino acids 363–393) are essential for nucleolar localization in p53 protein.37–39 But other studies thought that the C-terminal of p53 deleting the last 30 amino acid could enhance the specific binding capacity of DNA of the core area40–42 and amino acids 339–346 represents the minimal p53 repression domain. 43 Our results support the viewpoint of the latter. In this study, we removed amino acids 357–393 and reserved amino acids 339–346. They have found that the COOH-terminal region (339–346) containing eight amino acids was essential for the activity of transcriptional inhibition. This minimal domain is exactly located on the E6-binding region, which may possibly affect transcriptional activity. Mutants of the eight amino acids exhibited loss of their repression activity. This minimal domain may directly hinder the transcriptional activity domain of the NH2 terminus and interfere with the transcriptional complex to achieve repression. 43
In conclusion, p53(DEL) may affect proliferation and invasive potential of human lung cancer cells. p53(DEL) can induce cell apoptosis and inhibit EMT occurrence in lung cancer cells. These characteristics were reverted by the treatment of with PFT-a, a p53 inhibitor. So p53(DEL) is a promising target for prevention and treatment of lung cancer.
Footnotes
Acknowledgements
The authors thank Jinzhao Li for technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The financial support was received from Beijing Natural Science Foundation (No. 7153162).